
Abstract
Nitrate reductase catalyses the oxidation of NAD(P)H and the reduction of nitrate to nitrite. NR serves as a central point for the integration of metabolic pathways by governing the flux of reduced nitrogen through several regulatory mechanisms in plants, algae and fungi. Bacteria express nitrate reductases that convert nitrate to nitrite, but mammals lack these specific enzymes. The microbial nitrate reductase reduces toxic compounds to nontoxic compounds with the help of NAD(P)H. In the present study, our results revealed that Bacillus weihenstephanensis expresses a nitrate reductase enzyme, which was made to generate the 3D structure of the enzyme. Six different modelling servers, namely Phyre2, RaptorX, M4T Server, HHpred, SWISS MODEL and Mod Web, were used for comparative modelling of the structure. The model was validated with standard parameters (PROCHECK and Verify 3D). This study will be useful in the functional characterization of the nitrate reductase enzyme and its docking with nitrate molecules, as well as for use with autodocking.
1 Introduction
Human excretion, agricultural activities and the industries producing fertilizer, meat and food, milk, and detergents are the main sources of nitrogenous wastes. The ground water nitrate (NO3) concentration has increased globally [Citation1,Citation2]. Nitrate is transformed into nitrite in the digestive system of humans, which causes the condition known as methemoglobinemia, also called Blue Baby Syndrome [Citation3]. Proper handling can prevent nitrate contamination in the ground water and soil, and if it is not handled correctly, it can create several hazards. The increase in the pollution of natural sources of drinking water demands proper attention for the development of technologies for water remediation [Citation4]. For traditional physical and chemical techniques, such as ion exchange, reverse osmosis, catalysis and electro-dialysis, the costs of operations are high, and the methods are inefficient in removing waste brines from large volumes of waste [Citation5,Citation6]. Biological denitrification (alternative technology) methods are efficient methods and produce no secondary waste products [Citation7,Citation8]. Yan et al. [Citation9] reported that in aquatic systems, nitrate and nitrite can be reduced to ammonium, which can then be used for the synthesis of amino acids and the regulation of other biological pathways.
Table 3 Ramachandran plot analysis for modelled protein (nitrate reductase).
Table 4 No. of helices and turns in nitrate reductase enzyme.
The last step of complete heterotrophic denitrification is the final conversion of nitrogen gas (N2), which is made by the sequential reductive reactions of nitrate (NO3) to nitrite (NO2), nitric oxide (NO) and nitrous oxide (N2O). Enzymes such as nitrate reductase (Nar), nitrite reductase (Nir), nitric oxide reductase (Nor) and nitrous oxide reductase (Nos) play a role in this process [Citation10].
Cupriavidus necator produces periplasmic nitrate reductase (NapAB), a heterodimeric protein that belongs to the dimethyl sulfoxide reductase family of mononuclear Mo-containing enzymes and catalyses the reduction of nitrate to nitrite. The nitrate reductase from Desulfovibrio desulfuricans ATCC 27774 (DdNapA), a monomeric protein of 80 kDa, harbours a bis(molybdopterin guanine dinucleotide) active site and a (4Fe-4S) cluster. DdNapA was co-crystallized with its substrates and inhibitors, and the corresponding structures were solved at resolutions ranging from 1.99 to 2.45 Å [Citation11]. Einsle [Citation12] stated that the final product of an ammonium ion was produced and bound in coordination with the substrate nitrite to the active site of the heme iron, though the free electron pair at the nitrogen atom was reduced by the transfer of an electron and a proton in the series. While no intermediate reactions were found, NrfA was able to reduce various other nitrogen oxides, such as nitric oxide (NO), hydroxylamine (H2NOH) and nitrous oxide (N2O). Sulfite is the only known direct link between the nitrogen and sulfur cycles.
The molecular docking of the protein was performed to directly compare the predicted data with the experimental data and to determine the potential modes of action of the nitrate reductase enzymes. The combined data help us to understand the structure–activity relationships between the selected proteome and the binding sites for the nitrate molecule. Most protein families have three-dimensional structures, and it is likely to identify a homolog of a known structure from sequence database searches. Entrez's 3D-structure database has made it easy to access structural information and the functional annotation of proteins. Gavanji et al. [Citation13] showed that the important cellular enzyme nitrate reductase (Nar) plays a major part in cell activity. In the present study, the DiANNA 1.1 web server, Molegro Virtual Docker (MVD) and MetalDetector v2.0 software were used. The results obtained from docking showed the best pose, which was derived from the MolDock score for Catalase-peroxidase 30.7299 with a re-ranking score equal to 35.1088. However, a study by Georrge and Umrania [Citation14] utilized an in silico-based approach for the identification of drug targets. A comparison of the proteomes of the causal organism and humans was made to screen out non-homologous proteins. Different databases were used to find novel drug targets, and various tools were used for the prediction of sub-cellular localization and membrane proteins.
In the present study, an effort was made to generate the three-dimensional (3D) structure of the nitrate reductase (A9VSW1, A9VSW2, A9VSW3 and A9VSW4) from Bacillus weihenstephanensis. Six different methods, namely Phyre, RaptorX, M4T Server, HHpred, SWISS MODEL and Mod Web, were used for the comparative modelling. The model was validated with standard parameters (PROCHECK and Verify 3D).
2 Materials and method
2.1 Software and hardware
Automated comparative modelling was performed by Phyre 2 [Citation15], RaptorX [Citation16], M4T Server [Citation17], HHpred [Citation18], SWISS model [Citation19] and Mod Web [Citation20], and the resulting models were evaluated by PROCHECK [Citation21] and Verify 3D [Citation22]. Docking studies were carried out in the Autodock V 4.0 Workspace [Citation23]. Interactive visualization and analyses of the molecular structures were carried out in Pymol Viewer.
2.2 Analysis of proteome for Bacillus weinstephnisis (DS45)
The genomes of the effective nitrate-reducing bacterial strains DS45 were sequenced by 16S rRNA using NCBI-BLAST. The genome sequences of the DS45 strains were similar to the dissimilatory nitrate reducer B. weihenstephanensis. The proteome of B. weinstephnisis (Taxon Identification: 272620) was downloaded from UniProt (www.uniprot.org). The B. weihenstephanensis proteins were analyzed using CD-HIT to identify the paralogous or duplicate proteins. The sequence identity cut-off was kept at 0.6 (60% identity), and the global sequence identity algorithm was selected for alignment of the amino acids. A bandwidth of 20 amino acids and default parameters for alignment coverage were used. These proteins were subjected to screening for fragment sequences and were built using multiple sequence alignments, phylogenetic analysis, comparative modelling and docking (Autodock, which docks a ligand into a crystal structure using a Lamarkian genetic algorithm, was used) with the nitrate molecules.
2.3 Multiple sequence alignment
The sequence analysis of both bacterial strains was carried out using the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/). A multiple sequence alignment of the nitrate reductase of B. weinstephnisis with those of other bacterial strains was done using multiple sequence alignment tools (Clustal W).
2.4 Phylogenetic analysis
Acquired sequences were used for a gene homology search with the 16S rDNA sequences available in the public databases from BLAST [Citation24] and were identified to the generic level. Using the CLUSTAL-W Multiple Sequence Alignment Program, the 16S rDNA sequences of the isolated strains were aligned with the sequences of related organisms obtained from GenBank. Phylogenetic analysis was performed using the Bio Edit and Mega software programs, and a phylogenetic tree was constructed with the neighbour-joining method using the Tree View Program. To validate the reproducibility of the branching pattern, a bootstrap analysis was performed.
2.5 GenBank accession number
The partial 16S rDNA sequences of the bacterial strain B. weinstephnisis (DS45) were deposited in GenBank under the accession number KF926418.
2.6 Comparative modelling
An effort was made to generate the three-dimensional (3D) structure of the nitrate reductase (A9VSW1, A9VSW2, A9VSW3 and A9VSW4) from B. weinstephnisis. In brief, searches for sequence similarity within the BLAST database were performed with the BLAST program. The Psi-Blast sequence alignment program was used to find the template. The resulting sequences were aligned using the Clustal W software in the default set-up. There are several methods and computer programs for protein comparative modelling, such as Phyre2, RaptorX, M4T Server, HHpred, SWISS MODEL and ModWeb, and these programs were used with the respective templates downloaded from the Brookhaven PDB using a comparative modelling approach. The steepest descent energy minimization was done using the GROMOS 96 force field to regularize the structure geometry of the model [Citation25]. The final model was validated using the Swiss-Model Assessment Server for PROCHECK and Verify 3D.
2.6.1 SWISS-MODEL
Swiss-Model is an automated server that uses the sequence of a target protein to predict its structure. The purpose of this server is to make protein modelling accessible to all biochemists and molecular biologists in the world [Citation26]. A personal working environment is provided for each user where several modelling projects can be carried out in parallel. Tools for template selection, model building and structure quality evaluation can be utilized within the workspace. This server is capable of storing a user job for only 14 days [Citation27].
2.6.2 MODWEB
ModWeb is a web server for automated comparative protein structure modelling [Citation28]. MODWEB accepts one or many sequences in FASTA format [Citation29] and calculates models for proteins based on the best available template structures in the Protein Data Bank [Citation30]. Alternatively, MODWEB also accepts a protein structure as the input and calculates models for all of its identifiable sequence homologs in the non-redundant SWISS-PROT protein sequence database [Citation31]. The latter mode is a useful tool for various structural genomics efforts to assess the impact of a newly determined structure on the modelling coverage of the sequence space [Citation32,Citation33].
2.6.3 HHpred
The HHpred server was applied to fill the gap in the fast and widely used homology search programs, such as BLAST, PSI-BLAST [Citation34] or HMMer/Pfam [Citation35], and it is very sensitive and accurate, but the server is rather inflexible and has slow automated protein structure prediction servers. Thus, HHpred is mainly meant to be used as an interactive function and structure prediction server, allowing for the searching of various databases, the manual selection of templates, or the correction of errors in the proposed target–template alignment. We participated in the CASP8 competition and used three fully automated versions to test the accuracy of HHpred.
2.6.4 RaptorX
Normally, homology modelling and protein threading methods use a single template-sequence alignment to generate a protein's structure, but if the sequence is distantly related to the template or if the template does not achieve complete sequence coverage, the resulting model would not be accurate or fold properly. These programs suffer from two major drawbacks: (1) because these programs use a linear scoring function to guide the alignment of the template with the sequence, they cannot address some of the correlations among the features of proteins, and (2) these methods cannot give an accurate alignment if the sequence profile is sparse [Citation36].
2.6.5 Phyre2
Phyre2 is a free online homology modelling server [Citation37,Citation38]. Phyre2 uses the alignment of hidden Markov models via HHsearch to significantly improve the accuracy of alignments and detection rates. This incorporates a new ab initio folding simulation called “Poing” to model the regions of proteins in question that have no detectable homology to known structures [Citation39].
2.6.6 M4T server
The M4T server performs three main tasks in an automated manner: (i) template search and selection performed by the Multiple Template (MT) module, (ii) target sequence to template structure(s) alignment performed by the Multiple Mapping Module (MMM) [Citation40], and (iii) model building performed by Modeller [Citation41].
2.7 Model refinement
The theoretical structure of nitrate reductase was analyzed for its stereochemical clashes by subjecting the model to analysis by the Swiss PDB Viewer. An energy minimization with a harmonic constraint of 100 kJ/mol/Å2 was applied to all of the protein atoms using the Steepest-Decent and Conjugate Gradient technique to eliminate bad contacts between protein atoms. The computations were carried out using GROMOS96 in the Swiss-PDB Viewer. The backbone conformation was evaluated by inspecting the Psi/Phi Ramachandran plot that was obtained from the PROCHECK analysis.
2.8 Ramachandran plot
A Ramachandran plot provides the residue position in a particular segment based on the φ and ψ angles between the C∞–C and N–Cα atoms of the residues, respectively [Citation42]. It also increases the numbers of known protein structures and accuracy in ultra-high-resolution protein structures. In the present study, a Ramachandran plot was used to analyse the model [Citation43].
2.9 Molecular docking
Docking was performed by the AutoDock 4.0 program using the implemented empirical free energy function and the Lamarckian Genetic Algorithm (LGA) [Citation23]. Standard values for all docking parameters were used as described previously [Citation23], except the amount of independent docking runs performed for each docking simulation, which was set to 200. Cluster analysis was performed on the docked results using a root mean square (RMS) tolerance of 0.5 Å, and the initial coordinates of the ligand were used as the reference structure. AutoDock predicts the bound conformations of a small, flexible ligand to an inflexible macromolecular target with a known structure. The protein structure and nitrate molecules (ligand) were docked using the tools found in AutoDock. The prepared molecules were submitted to the software and saved in a grid file format. Finally, the molecules were run using command terminal. The docked file was analyzed for the best score values and viewed by a visualization tool such as Pymol.
3 Results and discussion
3.1 Inference of physiology from the phylogeny of organisms
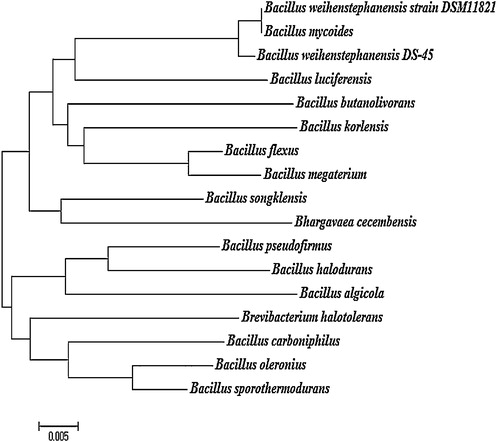
The evolutionary distance of B. weinstephnisis (DS45) was analyzed using phylogenetic classes. Phylogenetic analysis using the 16S rDNA sequences indicated that all of the bacterial isolates belonged to the genus Bacillus. From the branching pattern of the tree, the bacterial strain B. weinstephnisis was found to be closely related to B. luciferensis, B. butanolivorans, B. korlensis, B. flexus, B. megaterium, B. songklensis, Bhargavaea cecembensis, B. pseudofirmus, B. halodurans, B. algicola, Brevibacterium halotolerans, B. carboniphilus, B. oleronius and B. sporothermodurans (). The phylogenetic analysis showed that these bacteria formed an apparent cluster of nitrate reducers. Similarly, Musat et al. [Citation44] and Widdel et al. [Citation45] reported the “metabolic” clustering of anaerobic hydrocarbon degraders that were also degraders of higher alkanes and aromatic hydrocarbons, and these degraders include sulfate or nitrate-reducing bacteria, which have a common ancestor as explained by evolutionary divergence. Xu and Cote [Citation46] used the DNAPARS program of the PHYLIP package for the alignment and sequencing of the 220-bp sequences of the 40 Bacillaceae species. Therefore, the isolated strains were identified as Bacillus species based on their morphological, cultural, physiological, and biochemical characteristics as well as the 16S rDNA sequence analyses.
Fig. 1 Phylogenetic analysis of Bacillus weihenstephanensis (DS 45).
3.2 Proteome analysis for B. weinstephnisis (DS45)
Various enzymes present in the periplasm of several proteobacteria (alpha, beta, gamma and epsilon divisions) and the internal membrane, such as nitrate reductase (Nar), nitrite reductase (Nir), nitric oxide reductase (Nor) and nitrous oxide reductase (N2OR), catalyze denitrification reactions [Citation47]. One drawback of the molecular systems involved in this reduction is that they have been identified only partially at the protein level [Citation48]. When comparing the Gram-positive genomes of denitrifying organisms to Gram-negative organisms, the putative proteins have low DNA sequence homology, which may also imply the involvement of new genes in denitrification pathways, which may be the reason for overlooking the pathways [Citation49]. Hence, in this study, the nitrate reductase enzymes present in Archaea, Bacteria and Eukaryota organisms were analyzed. The numbers of sequences found in UniProt were 408, 55,338 and 2200, from which the Protein Data Bank confirmed the 3D structures of only 6 organisms in Archaea, 60 organisms in Bacteria (8 had nitrate binding protein domains), and 11 organisms in Eukaryota (). B. weihenstephanensis (Taxon Identification: 272620) consists of 6037 proteins, and these sequences were downloaded from the UniProt database [Citation50] (). The CD-HIT tool was used to identify the homologous proteins, and a 60% sequence identity was used as the threshold. Out of the 6037 proteins, 348 duplicate proteins were found. The remaining 5689 proteins were searched for nitrate reductase enzymes. Finally, four nitrate reductase proteins were identified as nitrate reductase alpha subunit (A9VSW1), nitrate reductase beta subunit (A9VSW2), nitrate reductase molybdenum co-factor (A9VSW3) and nitrate reductase gamma subunit (A9VSW4). The nitrate reductase alpha and gamma subunits were of close relation among the nitrate reductase enzymes compared to the beta and molybdenum cofactor enzymes, which had similar protein sequences during phylogenetic analysis (). The nitrate reductase enzyme of B. weihenstephanensis does not have any tertiary structure in the Protein Data Bank. Hence, this study predicts the structure and the docking of nitrate molecules with the help of Autodock tools.
Fig. 2 Phylogenetic analysis for nitrate reductase enzyme in Bacillus weihenstephanensis.

Table 1 Details of nitrate reductase enzyme present in sequence and structural database.
Table 2 Total number of nitrate reductase (Bacillus weihenstephanensis) in Uniprot.
3.3 Model building
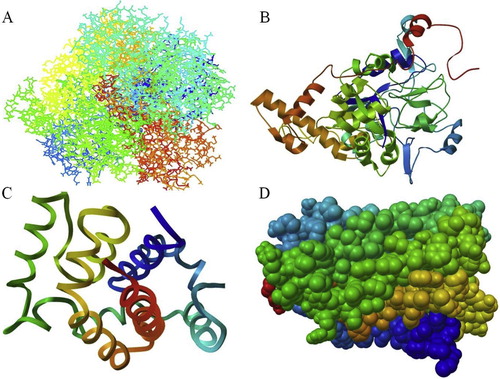
This study was performed based on the study of Seenivasagan et al. [Citation51]. The study used a comparative modelling approach to predict the structure of a VpR using the NMR structure of the HIV-1 regulatory protein as the template. The theoretical structure of VpR was generated using Modeller 9v1, a program for the comparative modelling of proteins using special restraints. Based on the results obtained from the different model prediction servers, the best-modelled structures for A9VSW1, A9VSW3 and A9VSW4 were achieved by the RaptoX server, while the best model of A9VSW2 was created by the M4T Server. The predicted structures were validated with a validation server (Procheck, WhatIF and Verify 3D). Energy minimizations were performed using Deep View with the GROMOS96 parameter, which does not significantly modify the initial models ().
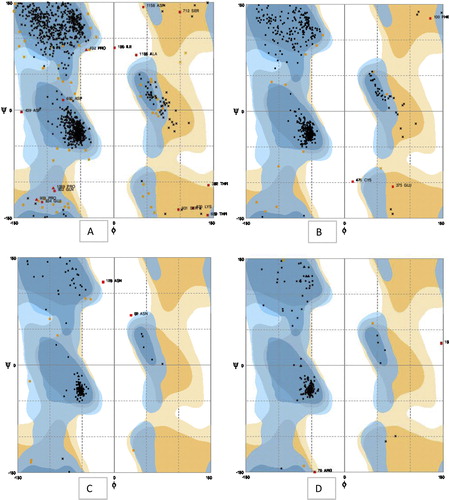
Fig. 3 Ramachandran plot analysis for (A) A9VSW1, (B) A9VSW2, (C) A9VSW3 and (D) A9VSW4 nitrate reductase.
3.4 Ramachandran plot analysis
Six comparative modelling servers were used to predict the protein structures. The modelled structures were screened for the number of residues modelled, and most of the residues were present in the favoured regions as shown by the Ramachandran plot. The theoretical structure of nitrate reductase was energy minimized and subjected to the PROCHECK web interface for stereochemical analysis () ( and ).
Fig. 4 Predicted structure for nitrate reductase enzyme: (A) A9VSW1, (B) A9VSW2, (C) A9VSW3 and (D) A9VSW4.
3.5 Analysis of docking results
PyMOL was used to generate docking poses by directly loading the docking programs through the plugin. The configuration/score relationships were analyzed directly in a small text box, which contains the docking score of the meta information. The ranking lists of the docked poses were prepared with the docking scores, which were used for exporting the docked ligands and their respective binding poses. Nitrate reductase alpha subunit (A9VSW1) showed that the protein/ligand formation was the best in the 4th (Binding Energy: −3.94) run, with a ligand hydrogen bond with LYS4:HN (1.802 Å), while beta subunit (A9VSW2) showed the best in the 10th (Binding Energy: −3.94) run, with the ligand hydrogen bonds with TRY452:HN (1.987 Å) and ARG455:HH11 (1.815 Å). Nitrate reductase molybdenum cofactor (A9VSW3) showed its best ligand formation in the 5th (Binding Energy: −5.15) run, with the ligand hydrogen bonds with HIS100:HD1 (2.022 Å), LYS103:HZ3 (1.991 Å) and LYS138:HZ2 (2.064 Å). The gamma subunit (A9VSW4) showed its best in the 2nd (Binding Energy: −5.52) run, with the ligand hydrogen bonds with LYS43:HN (2.245 Å), LYS44:HZ3 (2.06 Å) and ARG116:HH12 (1.97 Å). In all of the above, the resulting docking scores were ranked as 1 (; ). Similarly, Georrge and Umrania [Citation14] investigated potential drug targets in K. pneumonia using an in silico-based approach for the identification of drug targets. Different databases were used to find novel drug targets, and various tools were used for the prediction of the sub-cellular localizations of proteins and membrane proteins. Seeliger and Groot [Citation52] reported on an interface related to the popular molecular graphics system PyMOL and the molecular docking suites Autodoc and Vina, which provided structure-based drug design aided by the combination of docking and visualization.
Fig. 5 Docking structure for nitrate reductase enzyme: (A) A9VSW1, (B) A9VSW2, (C) A9VSW3 and (D) A9VSW4.

Table 5 Docking energy values.
4 Conclusion
B. weihenstephanensis was used as a structural model for the nitrate reductase enzyme in this study. The 3D structure of a protein is very important to understand its molecular functions. Structural analysis of selected nitrate reductase enzymes was done using different molecular modelling servers, including a specially designed server that provides even non-expert users with high quality comparative models in competition with those produced by manual expert modellers. The model was validated with standard parameters (PROCHECK and Verify 3D). The docking poses were ranked according to their docking scores, and the hydrogen bonds of docked ligands and their corresponding binding poses might be exported. Among the four nitrate reductase subunits, the gamma subunit (A9VSW4) showed the highest binding factor with 3 hydrogen bonds on the 2nd run, followed by alpha (A9VSW1), molybdenum (A9VSW3), and beta (A9VSW2) on their 4th, 5th and 10th runs, respectively.
Acknowledgement
The authors are thankful to the University Grants Commission (UGC) of New Delhi for providing the financial support for this research.
Notes
Peer review under responsibility of Taibah University.
References
- J.ChoiB.BatchelorC.WonJ.ChungNitrate reduction by green rusts modified with trace metalsChemosphere862012860865
- V.ReE.SacchiE.AllaisThe use of nitrate isotopes to identify contamination sources in the Bou-Areg aquifer (Morocco)Proc. Earth Plan. Sci.72013729732
- T.Y.K.ChanVegetable-borne nitrate and nitrite and the risk of methaemoglobinaemiaToxicol. Lett.2002011107108
- O.S.G.P.SoaresJ.J.M.ÓrfãoM.F.R.PereiraNitrate reduction in water catalysed by Pd–Cu on different supportsDesalination2792011367374
- M.ShrimaliK.P.SinghNew methods for nitrate removal from waterEnviron. Pollut.1122001351359
- K.A.KaranasiosI.A.VasiliadouS.PavlouD.V.VayenasHydrogenotrophic denitrification of potable water: a reviewJ. Hazard. Mater.18020102037
- M.A.GomezJ.Gonzalez-LopezE.Hontoria-GarcıaInfluence of carbon source on nitrate removal of contaminated groundwater in a denitrifying submerged filterJ. Hazard. Mater.8020006980
- B.MorenoM.A.GomezA.RamosJ.Gonzalez-LopezE.HontoriaInfluence of inocula over start up of a denitrifying submerged filter applied to nitrate contaminated ground water treatmentJ. Hazard. Mater.1272005180186
- H.YanW.HuangC.YanX.GongS.JiangY.ZhaoJ.WangY.ShiStructure and mechanism of a nitrate transporterCell Rep.32013716723
- Y.PanL.YeB.NiZ.YuanEffect of pH on N2O reduction and accumulation during denitrification by methanol utilizing denitrifiersWater Res.46201248324840
- S.NajmudinJ.Pablo GonzalezC.TrincaoA.CoelhoM.F.S.A.MukhopadhyayC.Nuno CerqueiraI.Carlos RomaoJ.G.MouraD.Jose MouraC.BrondinoJ.Maria RomaoPeriplasmic nitrate reductase revisited: a sulfur atom completes the sixth coordination of the catalytic molybdenumJ. Biol. Inorg. Chem.132008737753
- O.EinsleStructure and function of formate-dependent cytochrome C nitrite reductase NrfAMethods Enzym.496201110.1016/B978-0-12-386489-5.00016-6
- S.GavanjiH.A.AzizB.LarkiA.MojiriBioinformatics prediction of interaction of silver nitrate and nano silver on catalase and nitrate reductaseInt. J. Sci. Res. Environ. Sci.1220132635
- J.GeorrgeV.UmraniaIn silico identification of putative drug targets in Klebsiella pneumonia MGH78578Indian J. Biotechnol.102011432439
- http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index
- http://raptorx.uchicago.edu/
- http://manaslu.aecom.yu.edu/M4T/
- http://toolkit.tuebingen.mpg.de/hhpred
- http://swissmodel.expasy.org/
- http://modbase.compbio.ucsf.edu/ModWeb20-html/modweb.html
- R.A.LaskowaskiM.W.MacarthurD.S.MossJ.M.ThorntonProcheck-a program to check the stereochemical quality of protein structuresJ. Appl. Crystallogr.261993283291
- R.LuthyJ.U.BowieD.EisenbergAssessment of protein models with 3-dimensional profilesNature35619928385
- G.M.MorrisD.S.GoodsellR.S.HallidayR.HueyW.E.HartR.K.BelewA.J.OlsonAutomated docking using a Lamarckian genetic algorithm and an empirical binding free energy functionJ. Comput. Chem.19199816391640
- http://www.ncbi.nlm.nih.gov/BLAST
- R.G.BodadeS.D.BeedkarA.V.ManwarC.N.KhobragadeInt. J. Biol. Macromol.472010298303
- http://swissmodel.expasy.org/workspace
- N.GuexM.C.PeitschSWISS-MODEL and the SwissPdb Viewer: an environment for comparative protein modelingElectrophoresis1815199727142723
- R.SanchezA.SaliLarge-scale protein structure modeling of the Saccharomyces cerevisiae genomeProc. Natl. Acad. Sci. U.S.A.9519981359713602
- W.R.PearsonFlexible sequence similarity searching with the FASTA3 program packageMethods Mol. Biol.1322000185219
- H.M.BermanT.BattistuzT.N.BhatW.F.BluhmP.E.BourneK.BurkhardtZ.FengG.L.GillilandL.IypeS.Jainet al.The Protein Data BankActa Crystallogr. D: Biol. Crystallogr.582002899907
- B.BoeckmannA.BairochR.ApweilerM.C.BlatterA.EstreicherE.GasteigerM.J.MartinK.MichoudC.O’DonovanI.Phanet al.The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003Nucleic Acids Res.312003365370
- A.Sali100,000 protein structures for the biologistNat. Struct. Biol.5199810291032
- D.VitkupE.MelamudJ.MoultC.SanderCompleteness in structural genomicsNat. Struct. Biol.82001559566
- S.F.AltschulT.L.MaddenA.A.SchafferJ.ZhangZ.ZhangW.MillerD.J.LipmanGapped BLAST and PSI-BLAST: a new generation of protein database search programsNucleic Acid Res.25199733893402
- S.R.EddyProfile hidden Markov modelsBioinformatics141998755763
- J.PengJ.XuRaptorX: exploiting structure information for protein alignment by statistical inferenceProteins792011161171
- J.SodingProtein homology detection by HMM-HMM comparisonBioinformatics2172005951960
- L.A.KelleyM.J.SternbergProtein structure prediction on the Web: a case study using the Phyre serverNat. Protoc.432009363371
- I.A.MalikS.SharifF.MalikA.HakimaliW.A.KhanS.H.BadruddinNutritional aspects of mammary carcinogenesis: a case–control studyJ. Pak. Med. Assoc.4361993118120
- B.K.RaiA.FiserMultiple mapping method: a novel approach to the sequence to structure alignment problem in comparative protein structure modelingProteins632006644661
- A.SaliT.L.BlundellComparative protein modelling by satisfaction of spatial restraintsJ. Mol. Biol.2341993779
- M.YadavA.SinghS.RathaurE.LiebauJ. Mol. Graph. Model.282010435445
- http://mordred.bioc.cam.ac.uk/∼rapper/rampage.php
- F.MusatA.GalushkoJ.JacobF.WiddelM.KubeR.ReinhardtAnaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteriaEnviron. Microbiol.112009209219
- F.WiddelK.KnittelA.GalushkoAnaerobic hydrocarbon-degrading microorganisms: an overviewK.N.TimmisHandbook of Hydrocarbon and Lipid Microbiology2010SpringerBerlin Heidelberg, Germany19982021
- D.XuJ.C.CotePhylogenetic relationships between Bacillus species and related genera inferred from comparison of 39 end 16S rDNA and 59 end 16S–23S ITS nucleotide sequencesInt. J. Syst. Evol. Microbiol.532003695704
- S.R.PauletaS.Dell AcquaI.MouraNitrous oxide reductaseCoord. Chem. Rev.201010.1016/j.ccr.2012.05.026
- SuhartiS.de VriesBiochem. Soc. Trans.332005130
- I.VerbaendertN.BoonP.De VosK.HeylenSyst. Appl. Microbiol.342011385
- http://www.uniprot.org/uniprot/?query=organism:272620±keyword:181
- R.SeenivasaganR.KasimaniM.ParthibanR.KalidossP.ShanmughavelComparative modeling of viral protein R (VpR) from human immunodeficiency virus 1 (HIV1)J. Proteomics Bioinform.12008073076
- D.SeeligerB.L.de GrootLigand docking and binding site analysis with PyMOL and Autodock/VinaJ. Comput. Aided Mol. Des.242010417422