Abstract
Little is known about the processes that enable influenza A viruses to jump into new host species. Here we show that the non-structural protein1 nucleotide substitution, A374G, encoding the D125G(GAT→GGT) mutation, which evolved during the adaptation of a human virus within a mouse host, activates a novel donor splice site in the non-structural gene, hence producing a novel influenza A viral protein, NS3. Using synonymous 125G mutations that do not activate the novel donor splice site, NS3 was shown to provide replicative gain-of-function. The protein sequence of NS3 is similar to NS1 protein but with an internal deletion of a motif comprised of three antiparallel β-strands spanning codons 126 to 168 in NS1. The NS1-125G(GGT) codon was also found in 33 natural influenza A viruses that were strongly associated with switching from avian to mammalian hosts, including human, swine and canine populations. In addition to the experimental human to mouse switch, the NS1-125G(GGT) codon was selected on avian to human transmission of the 1997 H5N1 and 1999 H9N2 lineages, as well as the avian to swine jump of 1979 H1N1 Eurasian swine influenza viruses, linking the NS1 125G(GGT) codon with host adaptation and switching among multiple species.
Introduction
In the past century, most pandemics were caused by influenza A viruses (IAV).Citation1 Although the natural reservoir of IAV is aquatic birds, IAV expand their host rangeCitation2 through undefined adaptive processes involving mutation and genome reassortment.Citation3,Citation4 Over the years, the IAV have adapted to acquire the ability to cross the species barrier and thus have gained the capacity to infect, and cause disease in several new hosts including humans.Citation5 However, little is known about the genetic mutations and processes by which the IAV is able to efficiently jump among species.
Many well-documented host-switching events have occurred for the IAV, such as in 1979, when an avian H1N1 IAV switched to the swine hostCitation6 which established a new ‘Eurasian avian origin’ swine flu lineage within the Eurasian swine populations.Citation7,Citation8 In 1997, 18 instances of poultry adapted highly pathogenic avian H5N1 IAV switching hosts to humans occurred in Hong Kong, which resulted in unusually severe disease and the deaths of six individuals.Citation9,Citation10 Two years later in Hong Kong, two instances of poultry adapted low pathogenic avian H9N2 IAV switching hosts to humans resulted in hospitalization, but with normal clinical disease and survival.Citation11 However, neither the H5N1 nor H9N2 infections established sustained human-to-human transmission. The Eurasian avian to swine transmission was permanent and involved continued swine-to-swine transmission, whereas the H5N1 and H9N2 avian strains were not sufficiently adapted to humans to establish transmission in the new human host. However, both of the 1997 H5N1 and 1999 H9N2 IAV virus lineages must have possessed genetic features that allowed the first steps in host switching, which is infection of a novel host.
Recently, the study of experimental adaptation of a human H3N2 flu within a mouse host has revealed the acquisition of many positively selected mutations in the viral genome,Citation12,Citation13,Citation14,Citation15,Citation16 which include 11 coding mutations in the non-structural protein 1 (NS1) gene.Citation15 The NS genome segment encodes 2 genes, the NS1 protein from the full length transcript and the nuclear export protein (NEP) (also known as NS2), by pre-mRNA splicing.Citation17 NS1 is a multifunctional protein that plays key roles in inhibition of host immune responses and viral replication.Citation18 Specific NS mutations selected on mouse adaptation were also independently acquired in highly pathogenic viruses. For instance, 226I and 227K NS1 mutations are convergent with the 1918 H1N1 Spanish flu,Citation13,Citation15 and the F103L and M106I NS1 mutations, that have been shown to be adaptive,Citation12 have been previously selected in the highly pathogenic avian H5N1 and its precursor from low pathogenic H9N2 lineages.Citation12 Among 11 individual adaptive NS1 mutations, the nonsynonymous nucleotide substitution A374G inducing the D125G(GAT→GGT) mutation caused the greatest increase in viral replication and virulence in the mouse in vivo, which were associated with increased RNA polymerase activity, interferon antagonism, and protein synthesis in mouse cells in vitro.Citation13 This mutated codon was adjacent to the independently selected NS1 coding mutation, M124I (nucleotide A372G).Citation13,Citation15
IAVs use the host splicing machinery to alternatively splice the NS gene,Citation19 producing the NS1 protein from the unspliced transcript and the NEP protein from the spliced transcripts, and similarly for the M gene,Citation20 producing the M1 matrix protein from the unspliced transcript and the transmembrane ion channel M2 protein from the spliced transcript. However, it was shown that the NS1 protein plays a role in splicing of host mRNA, by inhibiting the accumulation of a spliced cellular mRNA,Citation21,Citation22,Citation23 without affecting the splicing of the NS gene.Citation24
In this study, we show the first reported case of IAV adaptation to a new host by the selection of a mutation, NS1 D125G(GAT→GGT), which introduced a new splice site into the NS gene, hence producing the novel NS3 protein by alternative splicing. In addition, we found, that the 125G(GGT) codon is mostly found in viruses that have recently switched to new host species.
Materials and methods
Cell lines and virus strains
Mouse kidney epithelial cells (M1), human lung epithelial cells (A549) and 293T cells were maintained as described previouslyCitation12 in minimum essential medium (MEM) or DMEM (for 293T) (Gibco; Life Technologies Corporation, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (HyClone; Thermo Fisher Scientific, Ottawa, Ont., Canada), penicillin (100 U/ml), streptomycin (100 g/ml) and 2 mM L-glutamine (Gibco; Life Technologies Corporation). All cells were incubated at 37 °C in 5% CO2. Viruses were grown as previously describedCitation12,Citation14 in 10-day-old specific pathogen-free embryonated chicken eggs (Canadian Food Inspection Agencies, Ottawa, Ont., Canada) at 37 °C for 2 days and stored at −80 °C.
Virus yield in cells and mice
Madin–Darby canine kidney (MDCK), M1 or A549 cells were washed twice with 1× phosphate-buffered saline (PBS) and infected with a multiplicity of infection (MOI) of 0.001 in the presence of L-1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK) trypsin (1–0.25 µg/ml) in triplicate. Supernatant were collected at 12, 24, 48 and 72 h post-infection (hpi). Plaque titres were determined on MDCK cells. Groups of 12 CD-1 mice were anesthetized with halothane and infected intranasally with 5×103 plaque-forming unit (pfu) in 50–100 µl virus diluted in PBS for each of the recombinant viruses. Virus yields were determined as described previously.Citation13
Ethics statement
All in vivo research was performed in accordance with the guidelines of the Canadian Council on Animal Care as described previously.Citation13
Plasmids and rescue of recombinant viruses
Virus rescue of HK-wild type(HK-wt) and NS mutant viruses was performed as previously described.Citation12,Citation14 The nucleotide sequence of the full length segment 8 for HK-wt and mutant NS1 and NS3 genes are included as FASTA files in Supplementary Table S1. The A/HK/1/68 NS3 gene cDNA was synthesized and inserted into the pLLB plasmid by Biomatic Corporation (Cambridge, Ont., Canada).
Protein and RNA synthesis
A549 and M1 cells were washed twice with PBS and infected with 100 µl recombinant viruses at an MOI of 3 (protein) or 2 (RNA). Cells were incubated at 37 °C for 30 min, and overlaid with 3 ml of 1× MEM. Cells were harvested at 8 hpi. Each experiment was performed in two or more biological repeats. A549 cells were infected with 100 µl recombinant viruses at an MOI of 5. After 8 hpi, the cells were pulse-labeled with 80 µCi/ml 35S cysteine and methionine for 1 h in cysteine and methionine free media (Gibco; Life Technologies Corporation), cell lysates were collected with 1× SDS sample buffer and assessed by SDS–polyacrylamide gel electrophoresis (PAGE) and autoradiography as previously described.Citation12
RNA analysis
Total RNA was isolated from infected cells using RNeasy Protect Mini Kit (QIAGEN Inc., Toronto, Ont., Canada). After reverse transcription using oligo-(dT)20 for viral mRNA or IAV universal primer for viral RNA (vRNA), real-time quantitative PCR analysis was performed using gene-specific primers (Supplementary Table S2), as previously described.Citation13 Each experiment was performed in three biological repeats and two technical replicates. Conventional reverse transcription (RT)-PCR was also performed using HK-NS forward and HK-NS reverse primers that were complementary to terminal regions of the NS1 genome segment for both viral mRNA and vRNA Table S2 followed by Sanger sequencing of cDNA products (StemCore Labs (OHRI), Ottawa, Ont., Canada).
Splice site prediction
wt and mutant NS gene sequences, from A/Hong Kong/1/1968 (H3N2) were run through ASSPCitation25 (Alternative Splice Site Predictor) using acceptor and donor site cut-off values of 7 and 9, respectively (http://www.es.embnet.org/~mwang/assp.html).
Human splice site
Consensus sequence of splice site is bases of alignment of known human donor and acceptor site sequences extracted from EID (Exon-Intron database).Citation26 Generation of sequence logos was done by WebLogo.Citation27
Protein immunoblot analysis
Infected cells were washed twice with cold PBS, and were collected with 500 µl of 1× SDS buffer. Equal volumes of protein samples were on 10% or 12.5% polyacrylamide-SDS gels for 1 to 2 h at 120 V followed by transfer into polyvinylidene difluoride membranes (Millipore Corporation Canada; Cedarlane, Burlington, Ontario, Canada) for 1 h at 20 V at room temperature. Membranes were blocked with 10% dry milk in Tris-buffered saline or 5% bovine serum albumin in Tris-buffered saline for 1 h at room temperature, and then incubated with primary rabbit antibodies, NS1 (Dr E. Brown, University of Ottawa) or actin (Sigma, St Louis, MO, USA), followed by incubation with specific horseradish peroxidase-conjugated secondary antibody (Sigma). Signal detection was achieved using ECL plus (Pierce Protein Biology Products; Thermo Fisher Scientific) according to the manufacturer’s instructions.
NS1 immunoprecipitation
NS1 immunoaffinity chromatography was performed using 5 μl rabbit anti-NS1 immune serum bound to 20 μl Protein G Dynabeads (Life Technologies Corporation). A549 infected with rHK-wt or rHK-NS-D125G cell lysate harvested 8 hpi were incubated with NS1 antibody-conjugated beads for 1 h at room temperature while rocking, then were washed with NP-40 lysis buffer. The beads were resuspended in 1× SDS buffer and boiled before analysis by SDS–PAGE electrophoresis. Gels were stained with Coomassie blue, and protein bands for NS1 and NS3 were cut for mass spectroscopy analysis (Advanced Protein Technology Centre (SickKids), Toronto, Ont., Canada).
Mass spectroscopy (MS)
Gel bands for NS1-wt, NS1 124I 125G and NS3 were reduced with 30 µl of 10 mM DTT in 50 mM ammonium bicarbonate (30 min at 56 °C) and alkylated with 30 µl of 100 mM iodoacetamide in 50 mM ammonium bicarbonate (15 min in the dark at room temperature) before in-gel digestion by submersion in 13 ng/μl trypsin (sequencing grade modified trypsin; Promega Corporation, Madison, WI, USA) for 3 h at 37 °C. The eluted peptides were loaded onto a 150 µm ID precolumn (Magic C18; Michrom Biosciences) at 4 µl/min and separated over a 75 µm ID analytical column packed into an emitter tip containing the same packing material. The peptides were eluted over 2 h at 300 nl/min using a 0 to 40% acetonitrile gradient in 0.1% formic acid using an EASY n-LC nano-chromatography pump (Proxeon Biosystems, Odense, Denmark). The peptides were eluted into a LTQ-Orbitrap hybrid mass spectrometer (Thermo-Fisher, Bremen, Germany) operated in a data-dependant mode using multistage activation. MS was acquired at 60 000 full width at half maximum resolution in the Fourier transform mass spectrometer and MS/MS was carried out in the linear ion trap. Six MS/MS scans were obtained per MS cycle. The raw data files were searched using Sequest (Thermo-Fisher Scientific) which was set up to search for NS1 from Influenza A virus (A/Hong Kong/1-1/1968(H3N2) Genbank accession number ACF22215), using a parent ion accuracy of 10 ppm and a fragment accuracy of 0.5 Da. A fixed modification of carbamidomethyl cysteine and variable modifications of phosphorylated serine, threonine and tyrosine residues, as well as oxidized methionine were considered in the search. MS spectra are available upon request.
Genomic and phylogenetic analysis
We retrieved all 18 237 NS gene sequence from NCBI’s Influenza Virus ResourceCitation28 on 17 March 2012, and identified the NS genes with a glycine at position 125 of the NS1 protein. Phylogenetic trees were constructed from the NS. Sequence data that were edited and analyzed by using the Geneious (version 5.5.6).Citation29 Phylogenies used PAUP (phylogenetic analysis using parsimony, version 4.0), with 1000 neighbor-joining bootstrap replicates.
Sequencing
The NS gene segment of the recombinant viruses were sequenced by Sanger sequencing and gene segments used in this study were deposited in the GenBank under the following accession numbers: CY103965 (NS-124I), CY103967 (NS-125G), CY103966 (NS-124I+125).
Mouse interferon-β (IFN-β) enzyme-linked immunosorbant assay (ELISA) assay
Monolayers of M1 cells were infected with each of the following rHK viruses NS1-wt, NS1-M124I, NS1-D125G and NS1-M124I+D125G at an MOI of 1 and overlaid with serum-free MEM. The cell supernatants were collected and tested for IFN-β production 24 hpi. Mouse IFN-β was titrated relative to mouse IFN-β standards by commercial ELISA as described by the manufacturer, PBL InterferonSource (Piscataway, NJ, USA) with subtraction of PBS mock-infected M1 cells control values.
Synthesis of NS1 and NS3 by cell-free transcription and translation
NS1 (wt and D125G(GAT→GGT)) and NS3 genes were inserted into the bidirectional pLLB plasmid for expression in vitro using the TnT T7 Quick-Coupled Transcription/Translation System (Promega, Thermo Fisher Scientific) in the presence of 10 µCi 35S-methionine and cysteine (Express 35S Protein Labeling Mix; Perkin Elmer, Waltham, MA, USA). The mix was incubated with 40 µl TnT master mix in a 50 µl total volume for 90 min in a 30 °C water bath then analyzed by SDS–PAGE and autoradiography.
Transfection of 293T cells
wt, mutant NS1 or NS3 plasmid (1 µg) was mixed with Lipofectamine 2000 (Life Technologies Corporation) according to the manufacturer’s instructions and incubated for 30 min at room temperature. The plasmids and the transfection reagent were added to a monolayer of 293T cells in a 35 mm plate. The transfection mix was replaced by Opti-MEM (Life Technologies Corporation) 24 h post-transfection. The cells were collected and assessed by SDS–PAGE and immnunoblotting against NS1 antibody, 48 hpi.
Statistical analysis
Statistical analysis was performed using Microsoft Office Excel 2007 software.
Results
Effect on viral growth
To identify the specific effects of the M124I(A372G) and D125G(A374G) mutations on virus biology, we generated recombinant IAV containing the wt NS, M124I, D125G or the double-mutant (M124I+D125G) genes in the A/Hong Kong/1/1968 (H3N2) genetic background. Various mouse adapted mutations in the NS1 proteins have been shown to increase viral growth in vitro and in vivo.Citation12,Citation13 To determine how the M124I and D125G NS1 mutations affect viral replication, we measured viral growth in different hosts; in vitro, in MDCK canine kidney epithelial cells, M1 mouse kidney epithelial cells, A549 human lung epithelial cells, and in vivo, in CD-1 mouse lungs (). NS mutant viruses grew to similar or slightly lower titres relative to wt in canine and human cells ( and 1). In contrast, growth in mouse cells showed a significant (P<0.05) increase in viral growth compared to wt (), where mutation D125G enhanced growth 102-fold, followed by the double and M124I mutants, with 41- and 2-fold increased yields respectively. Likewise, in vivo viral growth in mouse lungs showed increased growth for D125G and the double-mutant 1 day post-infection, with 20- and 16-fold increases relative to wt, respectively (). Thus, the D125G NS mutation in human IAV showed a gain of function for viral replication in mouse tissues.
Identification of the novel NS3 protein
To determine whether the adaptive NS mutation affects viral protein synthesis, we infected M1 and A549 cells with wt and NS mutant viruses and collected cell lysates for immunoblotting with anti-NS1 serum. Immunoblotting detected the 26 kDa NS1 protein and a second protein of 20 kDa () in cells infected with the 125G, as reported earlierCitation13 and the double-mutant. This indicated that the 125G mutation induced the smaller, 20 kDa protein. Combining the 125G with the 124I mutation increased the expression level of the 20-kDa protein.
Figure 2 Presence of 20 kDa (NS3) band. (A) Immunobloting for endogenous NS1 in A549 or M1 cells infected with the wt, M124I, D125G or the double-mutant (M124I+D125G) NS1 gene viruses (MOI 3) or mock infected with PBS, 8 hpi. (B) Immunoblots for endogenous NS1 in M1 cells infected with a mouse adapted A/HK/1/68-11-MA21-1 virus possessing the NS1 D125G(GAT→GGT) mutation (MOI 0.5), human wt NS1 virus (MOI 3) or mock infected with PBS, 8 hpi. (C) Effect of NS mutations on the rate of viral protein synthesis in infected A459 cells. A549 cells were infected (MOI 5) and pulsed for 1 h with S35 8 hpi. Cell lysate was collected and used for SDS–PAGE and autoradiography.
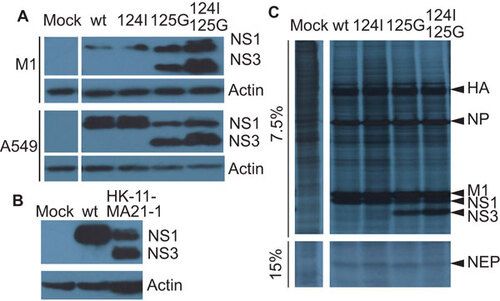
Furthermore, the 20-kDa protein was also detected in the original mouse adapted virus containing the 125G mutation (), and by radioactive labelling of proteins in infected A549 cells with the NS1 125G, and double mutant viruses (). The NS1 D125G and NS3 proteins are both stable in infected cells as previously shown in pulse labeling experimentsCitation13 (where the NS3 protein was initially identified as NS*).
We determined the amino acid sequence of the 20-kDa protein by liquid chromatography and tandem mass spectrometry (LC–MS–MS) of tryptic peptide fragments. NS1 and 20 kDa proteins were purified from wt and double-mutant-infected A549 cells by immuno-affinity chromatography and SDS–PAGE () for LC–MS–MS analysis. The extracted ion chromatograms confirmed the sequence of the wt and double mutant NS1 proteins ( and Supplementary Figure S1). In contrast, the 20-kDa protein sequence lacked three internal tryptic peptides () and possessed an additional unique ‘joining tryptic peptide’, with NS1 amino-acid 125G adjacent to 169H ( and Supplementary Figure S2). Thus, by peptide sequencing we identified the 20 kDa band as a novel influenza A protein, NS3, similar to NS1 with an internal deletion of 43 amino acids spanning positions 126K and 168G.
Figure 3 Characterization of NS3 by mass spectrometry. (A) Workflow for the production and purification of NS1 and 20 kDa (NS3) proteins for LC–MS–MS sequencing. (B) Identification, relative abundance, and sequence of extracted ion chromatogram peaks of NS1 wt or mutant tryptic peptides found in the NS1 and NS3 protein. (C) Relative abundance of extracted ion chromatogram peaks of a joining tryptic peptide with NS1 amino acids 125 bonded to 168. The MS/MS spectrum for fragment ions of NS3 joining peptide is included in Supplementary Figure S2. Approximate position of tryptic peptide relative to NS1 and NS3 is shown on schematic maps of the NS1 and NS3 proteins. Sequence coverage is included in Supplementary Figure S1.
The NS3 protein is a product of alternative splicing due to the A374G(125G) mutation
To determine whether the internal deletion of NS3 was due to splicing of the NS gene transcript, we sequenced NS RNA from NS wt, 125G and double-mutant infected M1 cells. Extracted RNA was reverse-transcribed using NS gene primers to generate cDNA from mRNA and viral genomic RNA. cDNA was amplified using PCR primers complementary to the terminal regions of the NS genome segment. At the mRNA level, the wt and all mutants amplified two bands encoding NS1 and NEP (). The 125G and double-mutant also amplified two novel bands of intermediate size. DNA sequencing showed that the smaller band corresponded to a novel transcript, with an internal in-frame deletion of 129 nucleotides spanning positions 374 and 502, involving predicted amino acids 125–168 ( and Supplementary Figure S3). This corresponded with the internal deletion found at the protein level in NS3. The larger novel band had a sequence consistent with a DNA hybrid of NS1 and NS3.Citation30 The 5′ end sequence of NS3 mRNA ends with the nucleotide preceding the mutation in codon G125 that was joined in-frame to position 2 of codon G168 which is the NEP 3′ splice acceptor site. This suggests that the formation of the NS3 transcript and protein is due to alternative pre-mRNA splicing of the NS transcript.
Figure 4 The NS1 D125G(GAT→GGT) mutation results in the formation of a novel splice product encoding NS3. (A) NS gene vRNA and mRNA from wt, M124I, D125G or the double-mutant (M124I+D125G) NS1 gene containing viruses were extracted from infected M1 cells (MOI 3) and amplified as cDNA by RT-PCR and were separated by agarose gel electrophoresis. Asterisk denotes bands corresponding to a NS1–NS3 hybrid artefact. (B) Schematic representations of NS transcripts, (NS1, NS3 and NEP), identified by sequencing of the PCR cDNA products. (C) Location of predicted alternative splice sites of wt, M124I, D125G and double-mutant (M124I+D125G) NS genes. (D) Nucleotide sequences of predicted splice site with likelihood scores (for sites in panel C) and resulting splice products (NEP or NS3) is shown for the NS1 wt, D125G and double-mutant (M124I+D125G). Human donor and acceptor splice site consensus sequences are shown above the splice sites of the NS genes. Position of the 124I and 125G mutations are highlighted in yellow and green respectively. (E–G), Levels of mRNA and vRNA. Quantitative RT-PCR was performed on total RNA from M1 cells infected with NS1 wt or mutant viruses (MOI 2), (n=3). (E) mRNA levels of NP, M1, NS1, NEP genes are shown relative to NS1-wt. (F) mRNA level of the NS3 transcripts. ND, Not detected. (E) vRNA levels of the NP, M, NS gene. (E, G) Results were normalized to β-actin levels, and presented as values relative to wt RNA levels. (E) Results were normalized as values relative to β-actin levels. (*P<0.05; **P<0.01; ***P<0.001; P value was determined by two-tailed student t with equal variance, (E,G) comparing the mutants to wt, (F) comparing the 125G to the double-mutant. Error bars indicate s.e.m.)
The RNA sequences of the various NS genes were analyzed using an algorithm to identify and predict potential human splice sites.Citation25 The wt and M124I mutant sequences showed one putative donor and acceptor splice site at position 30 and 503 () corresponding to the NEP splice sites. For the 125G and double mutation, an additional putative donor splice site was identified at position 373 corresponding to position 1 of pre-mRNA codon 125 (). The A to G mutation at position 374 forms a requisite GU donor splice site at the 5′ end of the intronCitation31 ( and 4), thus producing NS3 via the novel donor splice site at position 373 and the NEP’s acceptor splice site at position 503. Likelihood scores for the NS3 donor site were progressively higher than NEP. The addition of the 124I to the A374G(125G) mutation increased the likelihood score for the NS3 donor splice site (), since the 124I mutation further increased complementarity with the spliceosomal U1 snRNA binding sequence, ACUUACCU (identical in human and mouse), which initiates splicing.Citation31,Citation32
To assess the effect of the A374G(125G) mutation on RNA expression levels, RNA from infected M1 cells was quantified by quantitative RT-PCR for NS1, NEP, NS3, as well as a late (M1) and early gene transcript (NP). To specifically quantify the NEP and NS3 mRNA, we used primers for their respective donor splice sites; as for NS1, we used an intronic primer in the 125-168 area that is absent from the NEP and NS3 transcripts (Supplementary Table S1). The mutants showed a significant increases in mRNA and vRNA levels compared to the wt ( and 4), particularly in the double-mutant (with a greater than sevenfold increase for both mRNA and vRNA) that also affected the level of the NEP mRNA. The level of NS3 mRNA was undetectable for wt and 124I NS, and detectable in the A374G(125G) mutant with a 10-fold higher level in the double-mutant ( and Supplementary Figure S4). Thus, both mutations demonstrated a functional role in RNA synthesis. These data also demonstrated that increased similarity to the consensus sequence of the donor splice site due to the additional 124I mutation increases the splicing of the NS gene in favor of NS3.
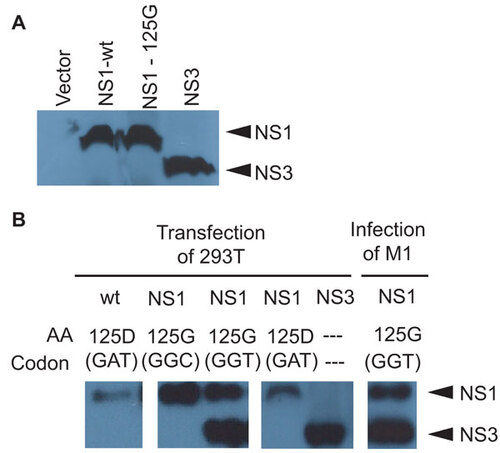
To further test the hypothesis that the novel 20-kDa NS3 band was the product of translation of a novel splice product of the NS gene, we expressed the NS1 D125G(GAT→GGT) mutant NS gene, as well as a synthetic cDNA construct of NS3 in splicing competent 293T cells and the splicing incompetent coupled prokaryotic T7 transcription/reticulocyte translation systems. Cell free transcription/translation of radioactively labeled NS1-wt and NS1 D125G(GAT→GGT) resulted in indistinguishable 26-kDa NS1 protein bands without the 20-kDa NS3 protein that was, however, synthesized from the synthetic NS3 cDNA construct (), confirming that the NS3 protein band was the product of this novel transcript. Expression of the synthetic NS3 cDNA in transfected 293T cells also resulted in the production of the 20-kDa NS3 protein band detectable by anti-NS1 immunoblotting that was also synthesized from the NS1 D125G(GAT→GGT) construct but not from the NS1 D125G(GAT-GGC) mutant (with the same NS1 coding sequence but with a defective splice donor site) (), indicating that NS3 protein production is dependent on splicing and furthermore is not the product of post-translational modification of the NS1 D125G mutant protein. Thus, the expression pattern of NS3 from various constructs in both eukaryotic and prokaryotic transcription systems supported the earlier findings that NS3 is the result of a novel spliced product of the NS1 gene. Further support that NS3 possessed a deletion of NS1 amino acids 126–168 was shown by the binding pattern of a novel panel of NS1 monoclonal antibodies that showed a lack of binding to NS3 for monoclonal antibodies that bind NS1 epitopes amino acids 138–147 and 161–169, whereas several other monoclonal antibodies, which bound outside the NS3 intronic region, were shown to bind both NS1 and NS3 (Rahim MN et al., unpublished).
Figure 5 Synthetic production of NS3. (A) In vitro expression of wt and 125G(GGT) NS1 and NS3 from coupled T7 transcription and reticulocyte translation of expression plasmids or empty vector in the presence of 35S labeled methionine and cysteine. Cell lysate was collected and used for SDS–PAGE and autoradiography. (B) Immunoblots for NS1 in transfected 293T cells with NS1 (wt, 125G(GGT) or 125G(GGC) mutants) or NS3 expression plasmids. Cell lysate was collected 24 h post-transfection. Control immunoblot is shown for endogenous NS1 and NS3 proteins in M1 cells infected with mutant NS1.
NS3 structural modeling
The deleted peptide in NS3 causes the loss of binding regions for the protein kinase R,Citation33,Citation34 cleavage and polyadenylation specificity factor,Citation35,Citation36 as well as a nuclear export sequence.Citation37 We used the known crystal structure of H5N1 NS1Citation38,Citation39 (Supplementary Figure S5) to illustrate the relative location of the deleted internal peptide region of NS3. The NS1 three-dimensional structure model shows that the internal deletion corresponds to a specific motif of NS1, composed of three anti-parallel β-strands.
Increase in viral growth is due to novel splicing of NS gene
To determine whether the initial observation of increased growth observed in mouse lungs and cells was due to the D125G(A374G) mutation in the NS1 protein or due to NS3, the novel splice product of the NS gene, we generated recombinant HK (A/HK/1/68) viruses with a glycine at position 125 of the NS1 protein, but lacking the novel NS3 donor splice site (). We generated two new recombinant viruses that contained a GGC or GGG codon at position 125, encoding a glycine but that cannot splice at this site due to the loss of the of the requisite GU 5′ donor splice site (). We measured growth, as performed in , in MDCK and M1 cells at 48 hpi and 1 day post-infection in CD-1 mice relative to wt and D125G(A374G) (). In MDCK cells the 125G(GGT) virus that produces NS3 protein grew to similar levels as the wt; however, the 125G(GGG) and 125G(GGC) viruses grew to significantly lower titres (480- and 18 000-fold less respectively) than the wt and 125G(GGT) viruses. In M1 cells and CD-1 mice, the 125G(GGT) grew to higher titres then the wt, as previously observed in (). Again, the 125G(GGG) and 125G(GGC) viruses grew to significantly lower levels than the 125G(GGT) mutant with 24- to 78-fold reduced titres in M1 cells, as well as significant reductions in CD-1 mice lungs (). Thus, the gain of function shown for viral replication in mouse tissues due to the D125G(GAT→GGT) NS mutation in human IAV was not due to the NS1 amino acid substitution at position 125 from aspartic acid to glycine, but was rather due to the novel splicing of the NS gene to produce NS3.
Figure 6 Effect of novel NS splicing on viral replication. (A) Schematic representations of the NS gene and its corresponding splice sites with predicted proteins from the NS1-wt, and NS3 splicing competent 125G(GGT), and splicing incompetent 125G(GGC) and 125G(GGG) mutants. (B) Immunobloting for endogenous NS1 and NS3 protein production in M1 cells infected with HK NS1 125G(GGT), 125G(GGC) and 125G(GGG) viruses or mock infected with PBS, 24 hpi showing an absence of NS3 protein for viruses without the NS3 donor splice site. (C) Virus growth in MDCK (MOI 0.001; n=3), M1cells (MOI 0.001; n=3) and in CD-1 mice (n=3) infected with influenza A viruses containing the either NS1-wt, NS3 splicing competent NS1 125G(GGT), or splicing incompetent 125G(GGC) and 125G(GGG) mutants. (**P<0.01; ***P<0.001; P value was determined by two-tailed student t with equal variance, comparing the mutants or wt to the 125G(GGT) at 48 hpi or 1 day post-infection. Error bars indicate s.e.m.)
To assess whether the increased replication seen in M1 cells in vitro was due to an increased ability to inhibit INF production (as previously shown in mouse lungsCitation13), we also measured IFN-β induction of infected cell supernatants using mouse IFN-β ELISA. Whereas HK-wt and NS1 M124I induced minimally detectable levels of IFN-β, the HK NS1 D125G(GAT→GGT) mutant produced significantly more IFN-β that was however reduced in combination with the M124I mutation (Supplementary Figure S6). Thus, the increased replication due to the NS1 D125G(GAT→GGT) in M1 cells cannot be explained by effects on inhibition of IFN-β induction. This was in contrast to the phenotype seen on infection of mice with NS1 D125G(GAT→GGT) where IFN-β induction was significantly reduced relative to HK-wtCitation13 indicating that IFN antagonism is a complex biological trait that differs for infections performed in vivo versus in vitro.
Bioinformatic analysis of 125G(GGT) NS1 codon in the influenza database
To have a better understating of the novel NS3 protein, we searched other NS1 gene sequences to assess the occurrence of the 125G(GGT) mutation among viruses found in the NCBI’s Influenza Virus Resource database. From the 18 237 available NS genes, most of the amino acids at NS1 position 125 were aspartic acid or glutamic acid (D or E) (Supplementary Figure S7 and Supplementary Table S3). Only 71 viruses had a glycine at position 125, with 33 viruses possessing the GGT codon required for the novel splicing of the NS gene needed to generate NS3 (). As only the RNA sequence GU (GT in DNA) is an absolute requirement in a splicing donor site, we also searched NS1 genes for the presence of codons ending in GT, including AGT, CGT, TGT, but none were found at position 125 of the NS1 protein.
Table 1 List of virus containing the NS1 125G(GGT) codon. Viruses are grouped according to phylogenetic origin
Figure 1 Host specific effects on viral replication. Virus growth in (A) MDCK , (B) A549, (C) M1cells (MOI 0.001, n=3) and (D) CD-1 mice (n=12) infected with influenza A viruses containing either the wt, M124I, D125G or the double-mutant (M124I+D125G) NS1 genes. (*P<0.05;**P<0.01; ***P<0.001; P value was determined by two-tailed student t with equal variance, comparing the mutants to wt 72 hpi or 1 day post-infection. Error bars indicate s.e.m.)
Although the pool of viruses containing the 125G(GGT) codon was small, a pattern emerged that associated this mutation with adaptation and host-switching (). 125G(GGT) was found in the first swine isolate of the wholly avian 1979 H1N1 virus (A/Swine/Belgium/WVL1/1979(H1N1)) that switched hosts from avian to swine in Europe (). NS1 125G(GGT) was also independently observed in one case of the 1997 H5N1 virus (A/Hong Kong/481/97(H5N1)) that switched hosts from avian to human and all (2 of 2) cases of the 1999 H9N2 viruses (A/Hong Kong/1074/99(H9N2) and A/Hong Kong/1073/99(H9N2)) that switched hosts from avian to human; and in the 2010 H3N2 virus that switch hosts from avian (A/Chicken/Korea/97/2007(H9N2)) to canine (A/Canine/Jiangsu/02/2010(H3N2))Citation40,Citation41 all of which linked the NS1 125G(GGT) mutation with interspecies transmission (). These natural observations are all in addition to the A374G (D125G) mutation which was also independently selected upon mouse adaptation of the human IAV (A/Aichi/2/68(H3N2)).Citation41
Furthermore the A/quail/Hong Kong/G1/97(H9N2) and A/Teal/Hong Kong/W312/97(H6N1) IAV which are proposed to be the viruses that rearranged by reassortment to generate the H5N1 and H9N2 influenza viruses that switched hosts from avian to human in Hong Kong in 1997 and 1999,Citation42,Citation43 also contain the 125G(GGT) codon (). This observation further strengthens the hypothesis that these quail and teal viruses were in fact the progenitors of the 1997 H5N1 and 1999 H9N2 virus. Viruses with the 125G(GGT) codon from the A/quail/Hong Kong/G1/97 lineages were isolated from a wide variety of avian species in Honk Kong, including pheasant, quail, pigeon, partridge and chicken ().Citation44 The NS gene with the 125G(GGT) codon from this lineage has been maintained in this lineage in poultry to be isolated 10 years later in domestic ducks in Vietnam (i.e. A/duck/Vietnam/OIE-2328/2009) ().
The recent canine IAV outbreak in KoreaCitation40 and southern ChinaCitation45 is hypothesized to be of avian origin from Korea due to the close phylogenic relationships.Citation46 Although the exact origin of this virus is still unknown, we found a closely related chicken virus from Korea (A/chicken/Korea/97/2007), which also contained the 125G(GGT) codon, which could be the source lineage of the canine IAV ().
In addition, two strain of H3N8 goose IAV from Zambia,Citation47 the one human H1N1 IAV from New York, and a pair of chickens infected with highly pathogenic avian H5N1 IAV from EgyptCitation48 also contained the 125G(GGT) mutation (), but were not associated or related with any known host switching event. The geographic location of the two H5N1 chicken IAV isolates from Egypt coincides with several accounts of human infection with avian H5N1,Citation48 however the human NS genes sequences were not available for comparative analysis.
Discussion
Here, we show that the A374G nucleotide substitution, encoding the D125G(GAT→GGT) mutation, selected upon lung serial-passage of human IAV within a mouse host, activates a new donor splice site, hence producing the NS3 mRNA transcript that encodes the novel NS3 protein.
Novel splice products where previously reported in different viruses for the M gene,Citation49,Citation50 but translation of the new mRNAs were not detected.Citation51 However, previous studies have shown that mutations that inactivate alternative splice sites in the M gene decrease the viruses viability and growth rate.Citation52 Viable genetic variants with different sizes of NS1 gene products due to premature stop codons have been reported on laboratory passage of pathogenic avian H7N3 IAV but these mutations attenuated growth in chickens.Citation53,Citation54 Although IAV variants possessing shorter length NS1 genes due to premature termination are common in natureCitation55 and have been generated by genetic engineering to produce attenuated vaccine candidates,Citation56 to the best of our knowledge, other than the NEP protein, no new splice products have been previously identified for the NS gene. Hence, the selection of an adaptive mutation introducing a functional splice site, and consequently the emergence of a novel intron which results in an in-frame deletion between the novel donor and previously described acceptor splice site constitutes the first-time alternative splicing has been associated with an adaptive gain-of-function in IAV.
When combining the 124I and 125G mutations, the level of NS3 mRNA transcription and protein synthesis increased, suggesting that the overall observed increases in NEP, NS1, M and NP vRNA and mRNA was controlled by higher levels of NS3 mRNA and/or protein production. The NS1 protein has been shown to regulate the splicing of IAV transcripts,Citation18 where it regulates the splicing of the M1 mRNA,Citation24 but not the splicing of the NS gene into NS1 and NEP.Citation24 However, the 124I mutation significantly increased splicing in favor of the novel NS3 protein when combined with the 125G mutation demonstrating an effect on splicing that may be due to changing the activity of the NS1 splice sites nonetheless, we cannot rule out an effect due to the 124I plus 125G mutated NS1 protein in this process.
By comparing growth properties of synthetic viruses with synonymous 125G mutations that were lacking the novel splice site, we were able to demonstrate that the observed increases in viral growth was primarily due to the novel splice product of the NS gene, and not due to the D125G amino acid change in the NS1 protein that resulted in decreased replication in M1 and MDCK cells (). We thus demonstrated increased replication of the splice competent NS1 D125G(GGT) mutant versus the splice incompetent D125G(GGC) and D125G(GGG) viruses that both grew to significantly lower yields in MDCK and M1 cells, as well as in mouse lungs. The 125G(GGT) mutation has also been shown to enhance IFN-β antagonism in the mouse lung that was associated with increased virulence and viral growth in the mouse lung.Citation13 This was in contrast to the increased IFN-β induction seen in M1 cells for the 125G(GGT) mutant (Supplementary Figure S6), indicating that IFN antagonism at the level of IFN-β induction is a complex biochemical trait. Forbes et al.Citation13 have also previously shown that the M124I mutation resulted in positive epistasis with the NS1 M106V mutation in mouse cells but negative epistasis in mouse lungs suggesting different types of NS1 gene interactions that are dependent on the host cell environment.
The increase in viral growth due to the (A374G)125G mutation was only detected in mouse, and not in human or canine cells. Taking into consideration that selection of the (A374G)125G mutation occurs when adapting a human IAV into a mouse host, this suggests that the increase in viral fitness due to the novel splice product, NS3, might only be beneficial in viruses that lack optimal functioning because they have not been adapted into a novel host. The (A374G)125G mutation in the human A/HK/1/68 virus in the human A549 cell was not required for replication in human cells and therefore did not enhance NS1 replicative functions, possibly because those functions have already been provided by previous human-adaptive mutations. Likewise, adaptive mutations such as the (A374G)125G did not enhance replication in the MDCK cell line, because the A/HK/1/68 virus possesses the appropriate functions of NS1 that are required to replicate in this cell line which is highly susceptible to infection with IAV from various host sources due, at least in part, to its defective IFN response due a defective Mx1 gene.Citation57 Thus, the 125G(GGT) mutations appear to be selected in viruses that have switched hosts and require mutations that compensate for a lack of adequate NS1 and/or NS2 gene functions in the new host. This hypothesis is supported by the natural occurrence of codon 125G(GGT) in avian viruses that have been adapted to and among various species of poultry, as well as poultry viruses that have gone on to transmit to mammals and thus were operational for influenza virus host switching. Thus in nature, NS1 125G(GGT) was found in several viruses that had switched hosts from avian to a mammalian, including human, swine and canine populations. Most of these events involved well characterized viruses, like the 1979 H1N1 virus progenitor for the Eurasian swine flu lineage, the 1997 H5N1 and the 1999 H9N2 virus that switched host from avian to human, or more recently for the 2010 H3N2 virus that switched hosts from avian to canine. In addition, the 125G(GGT) codon is also found in their progenitor viruses such as the A/quail/Hong Kong/G1/97 virus that established a prevalent Asian poultry lineage of H9N2, suggesting that the selection of the 125G(GGT) codon occurs on intensive adaption, such as occurs in high density poultry farming to increase the ability of a host switch in IAV. The accumulated data indicate that adaption to a new host is multigenic and involves changes in the properties of multiple genes (see Refs. 14–16) and therefore the 125G(GGT) codon would be expected to operate in host switching in concert with other adaptive mutations. The NS1 mutation (A374G)125G and predicted splice site was also independently selected upon mouse adaptation of the human IAV A/Aichi/2/68(H3N2),Citation41 providing further support that the novel splice product confers an adaptive advantage upon entry into a new host.
In this study, we established that the NS1 A374G substitution induced a D125G coding mutation and concomitantly activated a novel donor splice site, hence producing the novel viral transcript and protein, NS3. The NS3 splice site and resultant NS3 protein rather than the NS1 D125G substitution was critical for the adaptive properties due to this mutation. The increased viral replication and gene expression due to the new NS splice product and the A374G/D125G mutation, as well as antagonism of IFN-β production previously reported in mice,Citation13 provides a functional basis for examining the individual roles for the NS3 and NS1 125G proteins in viral RNA synthesis and IFN-β antagonism.Citation18 In addition, we observed an association of the 125G(GGT) with viruses that have switched hosts during both experimental and natural evolution. Although the NS3 splice site are similar in all 33 viruses with the 125G(GGT) mutation (Supplementary Figure S8), we are now investigating whether the novel splicing of the NS gene, and translation of the NS3 protein also occurs in natural viruses with the NS1 125G(GGT) codon.
The future occurrence of pandemics caused by the emergence of a novel influenza A virus against which the population has little or no immunity is unpredictable. The finding of the adaptive 125G(GGT) mutation improves our understanding of the adaptive roles of influenza A virus mutants and their properties that will open avenues for further research. Studies continue to find previously unknown IAV proteins such as PB1-F2,Citation58 and recently PA-X produced by frame shifting into the second open reading frame of segment 3.Citation59 Our finding suggests that the 125G(GGT) mutation that induces the NS3 protein from a novel splice product in human H3N2 virus, could be an important factor for the ability of other IAV to adapt to a new host, and hence, would be a good marker to identify the potential for the emergence of a novel pathogen in human, poultry, canine and swine populations.
Supplementary information
Download PDF (1,020.7 KB)Supplementary information
Download PDF (276.5 KB)Supplementary information
Download PDF (83.8 KB)We thank Shuzhi Wang (University of Ottawa, Ottawa) and Paul Taylor (The Hospital for Sick Children, Toronto) for technical assistance. This project has been supported by the CIHR Pandemic Preparedness Team grant to the CIHR Canadian Influenza Pathogenesis Team TPA-90188, and CIHR operating grant MOP-74526.
- Taubenberger JK, Morens DM. Pandemic influenza—including a risk assessment of H5N1. Rev Sci Tech2009;28: 187–202.
- Fields BN, Knipe DM, Howley PM. Fields virology. 5th ed.Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2007.
- Smith GJ, Vijaykrishna D, Bahl J et al.Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature2009;459: 1122–1125.
- Cauthen AN, Swayne DE, Schultz-Cherry S, Perdue ML, Suarez DL. Continued circulation in China of highly pathogenic avian influenza viruses encoding the hemagglutinin gene associated with the 1997 H5N1 outbreak in poultry and humans. J Virol2000;74: 6592–6599.
- Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe2010;7: 440–451.
- Pensaert M, Ottis K, Vandeputte J, Kaplan MM, Bachmann PA. Evidence for the natural transmission of influenza A virus from wild ducts to swine and its potential importance for man. Bull World Health Organ1981;59: 75–78.
- Donatelli I, Campitelli L, Castrucci MR, Ruggieri A, Sidoli L, Oxford JS. Detection of two antigenic subpopulations of A(H1N1) influenza viruses from pigs: antigenic drift or interspecies transmission? J Med Virol1991;34: 248–257.
- Vijaykrishna D, Smith GJ, Pybus OG et al.Long-term evolution and transmission dynamics of swine influenza A virus. Nature2011;473: 519–522.
- Claas EC, Osterhaus AD, van Beek R et al.Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet1998;351: 472–477.
- Chan PK. Outbreak of avian influenza A(H5N1) virus infection in Hong Kong in 1997. Clin Infect Dis2002;34 Suppl 2 S58–S64.
- Guo Y, Li J, Cheng X. [Discovery of men infected by avian influenza A (H9N2) virus.] Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi1999;13: 105–108. Chinese
- Dankar SK, Wang S, Ping J et al.Influenza A virus NS1 gene mutations F103L and M106I increase replication and virulence. Virol J2011;8: 13.
- Forbes NE, Ping J, Dankar SK et al.Multifunctional adaptive NS1 mutations are selected upon human influenza virus evolution in the mouse. PLoS One2012;7: e31839.
- Ping J, Dankar SK, Forbes NE et al.PB2 and hemagglutinin mutations are major determinants of host range and virulence in mouse-adapted influenza A virus. J Virol 2010; 84: 10606–10618.
- Ping J, Keleta L, Forbes NE et al.Genomic and protein structural maps of adaptive evolution of human influenza A virus to increased virulence in the mouse. PLoS One2011;6: e21740.
- Brown EG, Liu H, Kit LC, Baird S, Nesrallah M. Pattern of mutation in the genome of influenza A virus on adaptation to increased virulence in the mouse lung: identification of functional themes. Proc Natl Acad Sci USA2001;98: 6883–6888.
- O’Neill RE, Talon J, Palese P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J1998;17: 288–296.
- Hale BG, Randall RE, Ortin J, Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol2008;89( Pt 10): 2359–2376.
- Lamb RA, Lai CJ. Expression of unspliced NS1 mRNA, spliced NS2 mRNA, and a spliced chimera mRNA from cloned influenza virus NS DNA in an SV40 vector. Virology1984;135: 139–147.
- Lamb RA, Lai CJ. Spliced and unspliced messenger RNAs synthesized from cloned influenza virus M DNA in an SV40 vector: expression of the influenza virus membrane protein (M1). Virology1982;123: 237–256.
- de Vries W, Haasnoot J, Fouchier R, de Haan P, Berkhout B. Differential RNA silencing suppression activity of NS1 proteins from different influenza A virus strains. J Gen Virol2009;90( Pt 8): 1916–1922.
- Lu Y, Qian XY, Krug RM. The influenza virus NS1 protein: a novel inhibitor of pre-mRNA splicing. Genes Dev1994;8: 1817–1828.
- Fortes P, Beloso A, Ortin J. Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO J1994;13: 704–712.
- Robb NC, Jackson D, Vreede FT, Fodor E. Splicing of influenza A virus NS1 mRNA is independent of the viral NS1 protein. J Gen Virol 2010; 91( Pt 9): 2331–2340.
- Wang M, Marin A. Characterization and prediction of alternative splice sites. Gene2006;366: 219–227.
- Saxonov S, Daizadeh I, Fedorov A, Gilbert W. EID: the Exon-Intron Database-an exhaustive database of protein-coding intron-containing genes. Nucleic Acids Res 2000; 28: 185–190.
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res2004;14: 1188–1190.
- Bao Y, Bolotov P, Dernovoy D et al.The influenza virus resource at the National Center for Biotechnology Information. J Virol2008;82: 596–601.
- Meintjes P, Duran C, Kearse M et al.Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics2012;28: 1647–1649.
- Martins R, Silva B, Proenca D, Faustino P. Differential HFE gene expression is regulated by alternative splicing in human tissues. PLoS One2011;6: e17542.
- Ast G. How did alternative splicing evolve? Nat Rev Genet2004;5: 773–782.
- Carmel I, Tal S, Vig I, Ast G. Comparative analysis detects dependencies among the 5′ splice-site positions. RNA2004;10: 828–840.
- Li S, Min JY, Krug RM, Sen GC. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology2006;349: 13–21.
- Min JY, Li S, Sen GC, Krug RM. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology2007;363: 236–243.
- Chen Z, Li Y, Krug RM. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J1999;18: 2273–2283.
- Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3'end formation of cellular pre-mRNAs. Mol Cell1998;1: 991–1000.
- Li Y, Yamakita Y, Krug RM. Regulation of a nuclear export signal by an adjacent inhibitory sequence: the effector domain of the influenza virus NS1 protein. Proc Natl Acad Sci USA1998;95: 4864–4869.
- Bornholdt ZA, Prasad BV. X-ray structure of NS1 from a highly pathogenic H5N1 influenza virus. Nature2008;456: 985–988.
- Bornholdt ZA, Prasad BV. X-ray structure of influenza virus NS1 effector domain. Nat Struct Mol Biol2006;13: 559–560.
- Song D, Kang B, Lee C et al.Transmission of avian influenza virus (H3N2) to dogs. Emerg Infect Dis2008;14: 741–746.
- Narasaraju T, Sim MK, Ng HH et al.Adaptation of human influenza H3N2 virus in a mouse pneumonitis model: insights into viral virulence, tissue tropism and host pathogenesis. Microbes Infect2009;11: 2–11.
- Lin YP, Shaw M, Gregory V et al.Avian-to-human transmission of H9N2 subtype influenza A viruses: relationship between H9N2 and H5N1 human isolates. Proc Natl Acad Sci USA2000;97: 9654–9658.
- Guan Y, Shortridge KF, Krauss S, Webster RG. Molecular characterization of H9N2 influenza viruses: were they the donors of the ‘internal’ genes of H5N1 viruses in Hong Kong? Proc Natl Acad Sci USA1999;96: 9363–9367.
- Guan Y, Shortridge KF, Krauss S et al.H9N2 influenza viruses possessing H5N1-like internal genomes continue to circulate in poultry in southeastern China. J Virol2000;74: 9372–9380.
- Li S, Shi Z, Jiao P et al.Avian-origin H3N2 canine influenza A viruses in Southern China. Infect Genet Evol2010;10: 1286–1288.
- Lin Y, Zhao Y, Zeng X, Lu C, Liu Y. Genetic and pathobiologic characterization of H3N2 canine influenza viruses isolated in the Jiangsu Province of China in 2009–2010. Vet Microbiol2012;158: 247–258.
- Simulundu E, Ishii A, Igarashi M et al.Characterization of influenza A viruses isolated from wild waterfowl in Zambia. J Gen Virol2011;92( Pt 6): 1416–1427.
- Kayali G, Webby RJ, Ducatez MF et al.The epidemiological and molecular aspects of influenza H5N1 viruses at the human-animal interface in Egypt. PLoS One2011; 6: e17730.
- Shih SR, Suen PC, Chen YS, Chang SC. A novel spliced transcript of influenza A/WSN/33 virus. Virus Genes1998;17: 179–183.
- Shih SR, Nemeroff ME, Krug RM. The choice of alternative 5′ splice sites in influenza virus M1 mRNA is regulated by the viral polymerase complex. Proc Natl Acad Sci USA1995;92: 6324–6328.
- Jackson D, Lamb RA. The influenza A virus spliced messenger RNA M mRNA3 is not required for viral replication in tissue culture. J Gen Virol2008;89( Pt 12): 3097–3101.
- Chiang C, Chen GW, Shih SR. Mutations at alternative 5′ splice sites of M1 mRNA negatively affect influenza A virus viability and growth rate. J Virol.2008;82: 10873–10886.
- Perdue ML. Naturally occurring NS gene variants in an avian influenza virus isolate. Virus Res1992;23: 223–240.
- Wang L, Suarez DL, Pantin-Jackwood M et al.Characterization of influenza virus variants with different sizes of the non-structural (NS) genes and their potential as a live influenza vaccine in poultry. Vaccine2008;26: 3580–3586.
- Dundon WG. Variability among the neuraminidase, non-structural 1 and PB1-F2 proteins in the influenza A virus genome. Virus Genes2012;44: 363–373.
- Richt JA, Garcia-Sastre A. Attenuated influenza virus vaccines with modified NS1 proteins. Curr Top Microbiol Immunol2009;333: 177–195.
- Seitz C, Frensing T, Hoper D, Kochs G, Reichl U. High yields of influenza A virus in Madin–Darby canine kidney cells are promoted by an insufficient interferon-induced antiviral state. J Gen Virol2010;91( Pt 7): 1754–1763.
- Chen W, Calvo PA, Malide D et al.A novel influenza A virus mitochondrial protein that induces cell death. Nat Med2001;7: 1306–1312.
- Jagger BW, Wise HM, Kash JC et al.An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science2012;337: 199–204.