Abstract
Foamy viruses are complex retroviruses that have been shown to be transmitted from nonhuman primates to humans. In Bangladesh, infection with simian foamy virus (SFV) is ubiquitous among rhesus macaques, which come into contact with humans in diverse locations and contexts throughout the country. We analyzed microsatellite DNA from 126 macaques at six sites in Bangladesh in order to characterize geographic patterns of macaque population structure. We also included in this study 38 macaques owned by nomadic people who train them to perform for audiences. PCR was used to analyze a portion of the proviral gag gene from all SFV-positive macaques, and multiple clones were sequenced. Phylogenetic analysis was used to infer long-term patterns of viral transmission. Analyses of SFV gag gene sequences indicated that macaque populations from different areas harbor genetically distinct strains of SFV, suggesting that geographic features such as forest cover play a role in determining the dispersal of macaques and SFV. We also found evidence suggesting that humans traveling the region with performing macaques likely play a role in the translocation of macaques and SFV. Our studies found that individual animals can harbor more than one strain of SFV and that presence of more than one SFV strain is more common among older animals. Some macaques are infected with SFV that appears to be recombinant. These findings paint a more detailed picture of how geographic and sociocultural factors influence the spectrum of simian-borne retroviruses.
Emerging Microbes & Infections (2013) 2, e29; doi:10.1038/emi.2013.23
Introduction
The discovery that human immunodeficiency virus evolved from enzootic nonhuman primate (NHP)-borne viruses has stimulated an interest in other NHP-borne retroviruses and their transmission to humans.Citation1 Human societies critically influence the ecological contexts in which zoonotic transmission occurs.Citation2,Citation3,Citation4,Citation5 Nowhere is this more evident than in South and Southeast Asia, where the world’s densest human populations are situated close to some of the most diverse NHP fauna. Widespread destruction of NHP habitat, rapidly developing infrastructure, as well as social and economic forces, are changing the geographic landscape and bringing previously separate NHP populations into contact with each other, as well as into contact with humans. As a result, these new geographic relationships are likely to bring together various infectious agents harbored by these NHP populations, in new combinations, which may increase the potential for the emergence of zoonotic disease.Citation3,Citation6
At the epicenter of these forces lies Bangladesh, home to millions of people and thousands of NHP. The major river systems and its tributaries of Bangladesh are its most prominent geographic feature and have for centuries provided the means for vital trade and the movement of humans and their belongings throughout the Indian subcontinent.
The human population density in Bangladesh is among the highest in the world (https://www.cia.gov/library/publications/the-world-factbook/index.html). Most of the 161 million people live in crowded urban or peri-urban areas. While predominately a Muslim country, ∼10% of the population identify themselves as Hindu. Hindus consider NHP sacred, the ‘children’ of the monkey deity Hanuman. Because of these cultural/religious beliefs, NHP populations are often found in Hindu communities. Muslims takes a less sanguine view of monkeys, and since they are not hunted for bush meat, their presence is not always welcomed.Citation7
While Bangladesh contains a rich NHP fauna including five species of macaque (Rhesus (Macaca mulatta), Pig-tail (M. leonine), Stumptail (M. arctoides), Long-tail (M. fascicularis) and Assamese (M. assamensis) macaques) and three species of langur (Hanuman (Semnopithecus entellus), Capped (Trachypithecus pileatus) and Phayre’s langur (Trachypithecus phayrei)), only the rhesus macaque is still found in significant numbers though many of these free-ranging rhesus populations are now restricted to urban and peri-urban areas. With few exceptions the other NHP are found only in the isolated forest patches that remain along the borders with India and Myanmar.Citation8,Citation9 In addition to free-ranging rhesus populations, pet and performing monkeys can be found in Bangladesh. The latter are often associated with nomadic people found throughout South Asia (known as the Bade, or in other countries as Qalandar) who train these rhesus macaques to perform for audiences.Citation10 Relatively little is known about these people, their patterns of travel or their practices of animal husbandry.
Bangladeshi NHP come into contact with humans in several contexts: urban, shrine/temple, performing and wild, free-ranging populations in forested areas.Citation11,Citation12 Importantly, these NHP populations are not isolated from one another; they are dynamic and overlapping so that they can potentially intermix. Animals taken from the wild or urban areas become pets. Pets are released or escape into the wild. Abundant human/NHP contact makes Bangladesh a ‘natural laboratory’ for the study of cross-species transmission of infectious agents.Citation13
Bangladeshi NHP harbor at least three retroviruses. While simian betaretrovirus, previously known as simian retrovirus D (SRV-D), and simian T-cell lymphotropic virus (Jones-Engel, unpublished data) are found in only a few, isolated populations of some species, simian foamy virus (SFV) is ubiquitous. SFV is a member of a subfamily of retroviruses, the Spumaretrovirinae, which are distinct from other retroviruses by their mode of replication. For example, the orthoretroviruses such as human immunodeficiency virus are RNA viruses that reverse transcribe their genomes into DNA when they infect new cells. In contrast, spumaretroviruses are DNA viruses that package RNA and reverse transcribe their genomes during assembly and/or release. In this manner, they resemble the hepadnaviruses such as hepatitis B virus.Citation14 Foamy virus (FV) naturally infect cats, cows, some horses and all species of NHP tested. Phylogenetic analyses suggest that foamy-like viruses evolved over 400 million years ago, making them the oldest known animal viruses.Citation15 Although FV have been shown to be highly cytopathic in some cell types in culture, such as fibroblast lines, there is no evidence that they cause disease, either in their natural hosts, or in other mammalian species.Citation16 FV replicate only in the differentiating superficial epithelial cells of the oral mucosa in vivo and are efficiently secreted into saliva in natural hosts.Citation17,Citation18 However, most organs of FV-infected animals contain latent proviral DNA. Proviral DNA can readily be found in peripheral blood mononuclear cells.Citation19,Citation20 FV can be considered ‘perfect’ viruses in that they are transmitted very efficiently without inducing pathological changes that compromise the host.Citation14 Since essentially all adult NHP are infected with FV, it can be considered part of the normal host flora.
SFV is highly prevalent and is efficiently transmitted through saliva among rhesus macaques (up to 100% of free ranging macaques are infected by age 3).Citation21 Some human infections have been documented, but no human-to-human transmission has been reported.Citation22 As such, it is an appropriate agent for studying how parenterally transmitted viruses move within and between NHP populations, and from NHP to human populations. Several research groups have examined zoonotic transmission of SFV from NHP to humans. Zoonotic transmission has been documented in monkey handlers and laboratory technicians and veterinarians, bush meat hunters in Africa and temple workers in South and Southeast Asia that live near monkeys.Citation23,Citation24,Citation25,Citation26,Citation27
In the present study, we initially screened for SRV-D and SFV; we then characterized SFV sequences from 164 rhesus macaques from six sites (four urban and two forested) in Bangladesh, as well as from performing monkeys that were sampled at several locations around the country. The differences in the SFV gag gene present in different populations allowed us to delineate strains. We used sequence analysis to investigate the following questions: (i) can rhesus macaques be infected with more than one SFV strain at a time? (ii) do SFV strains recombine in naturally infected rhesus macaques? and (iii) do both natural geographic barriers and human influences contribute to SFV strain variation among Bangladesh rhesus macaques? Our data contribute to an understanding of the ecology of SFV in macaques at the human/primate interface. Further, when considered in conjunction with our accompanying paper for this project (Engel et al., in review), which characterizes the zoonotic transmission of SFV in these populations, our results enhance our understanding of the viral genetic factors that can influence zoonotic transmission.
Materials and methods
Ethics statement
These research protocols have been reviewed and approved by the University of Washington Institutional Animal Care and Research Committee (4233-01). This research adhered to the legal requirements of the countries in which the research was conducted. Biological samples were shipped internationally under a Convention on International Trade in Endangered Species permit.
Sampling sites
In the present study, free-ranging populations of rhesus macaques were sampled in six geographically distinct areas of Bangladesh as shown in Figure 1. These six sites were selected following a country-wide survey designed to locate NHP populations that were synanthropic (species that thrive in human-altered environments).Citation28 Dokhola is a protected forest area in north central Bangladesh. Permanent human establishments are few in this 8500-hectare preserve, though the area is heavily used by villagers living on the periphery, who collect resources from the forest. It is estimated that at least several hundred rhesus macaques in two or three large groups range throughout the protected area. Bormi and Dhamrai are densely populated urban areas located 150 and 100 km respectively south of Dokhola. These two urban areas are approximately 50 km apart. No natural patches of forest remain in these urban areas, though isolated large trees and numerous fruiting trees and bushes are maintained by local people for cultural and economic benefits. Both Bormi and Dhamrai have substantial Hindu populations, which are concentrated in specific locations. Many of the macaque sleeping trees are found within these Hindu areas, though the monkeys range throughout the urban sites.
Narayanganj is a major port city located southeast of the capital, Dhaka. The macaque population in this area has dwindled significantly over the past 5 years as the human density has increased substantially, resulting in nearly complete loss of any large trees. Dokhola, Bormi, Dhamrai and Narayanganj are all located northeast of the Padma River. This river is a formidable geographic barrier to natural macaque migration.
Charmaguria is located southwest of the Padma River. This urban site still contains a large number of forested areas that can and do support a robust population of macaques. Karamjal, also southwest of the Padma River, is a forestry station located at the northern tip of the Sundarbans, the world’s largest mangrove forest. This station is one of the tourist entry points into the Sundarbans and a small troop of rhesus macaques has become habituated to visitors at this site. The Sundarbans are prime habitat for rhesus macaques, though there are no comprehensive census data for macaques in this area estimates range to the thousands.Citation28
The nomadic Bade with their performing macaques were found as we traveled between sampling sites and during a visit to a village in western Bangladesh that is used as an intermittent base for the Bade. The Bade are readily identifiable by the distinctive design of their tents, usually erected by major roadsides on the edges of urban areas, together with the specially crafted wooden boxes that contain their performing monkeys. Figure 1 indicates the range in which we encountered the performing monkeys and their nomadic owners.
Figure 1 Rhesus macaque sampling site locations including relative geographic proximity and historical and extant forest patches. Rhesus macaques, Macaca mulatta, were sampled from six geographically distinct sites in Bangladesh (n=126). Concentric circles in 10 km intervals from the epicenter of the sites are shown. Existing forest areas are noted in dark green and historical forest cover is shown in lighter green. Natural macaque migration would have been possible between sites that were connected by forested areas. Nomadic Bade people travel throughout the subcontinent with their performing macaques. We sampled 38 of these performing monkeys from multiple locations within the shaded light blue area.

Sample collection from free-ranging monkeys in Bangladesh
Our group has trapped, darted, sampled and released free-ranging rhesus macaques (M. mulatta) in Bangladesh since 2006. Capture and sampling methods are detailed in Jones-Engel et al.Citation29 Rhesus trapping, darting and sampling only took place after we had met with village stakeholders and explained our proposed project and long-term objectives. We only sampled at sites where we had support of the local residents. At sites where we could identify more than one group of rhesus macaques, we attempted to obtain samples from individuals from each group. Briefly, free-ranging macaques were trapped in portable cages (measuring approximately 2.5 m3) fabricated at the Washington National Primate Research Center. Captured macaques were hand-injected intramuscularly with 3 mg of Telazol/kg of estimated body weight. All anesthetized macaques were given a physical exam. While under anesthesia, we performed morphometric analysis and collected biological samples. Using sterile technique and universal precautions, 5–10 mL of whole blood was collected via venipuncture from the femoral vein and stored in ethylenediaminetetraacetic acid tubes (VWR, Radnor, PA, USA) on cold packs. A portion of the whole blood was aliquoted and the remaining sample centrifuged to separate plasma from the cells and then aliquoted. Blood samples were kept on cold packs and then transferred to long term −80 °C storage. Each macaque’s dental formula was recorded for age assessment and animals were classified as ‘adult’ versus ‘juvenile’ based on the observed tooth eruption sequence. Each animal was given a unique identifier via a subcutaneous Avid microchip (Norco, CA, USA), to allow future longitudinal sampling. Following sample collection, the anesthetized animals were placed in recovery cages and allowed to recover fully before being released as a group back into their home range.
The same anesthetic and biological sampling protocols were used for animals that were darted and for the performing monkeys. We darted animals using the Telinject (Agua Dolce, CA, USA) blowpipe system. To minimize risk of injury, young animals and pregnant females were not darted. Prior to anesthetizing and sampling the performing monkeys, we discussed the procedures and our research objectives with the monkeys’ owners and collected information on the age of the animal and where it was originally trapped.
Microsatellite analyses
Microsatellite assay
Genomic DNA was extracted from 200 µL aliquots of whole blood collected in ethylenediaminetetraacetic acid using the QIAamp DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany). Animals that had tested positive for SFV were assayed using a 13-autosomal microsatellites panel (D01S548, D03S1768, D05S1457, D06S501, D07S794, D08S116, D09S921, D04S2365, D10S611, D11S2002, D13S765, D15S823 and D18S537).Citation30 Polymerase chain reaction (PCR) was carried out in a 15 µL volume reaction using 25–35 ng of total genomic DNA, 0.5 mM of each set of primers and 7.5 µL of 2X master mix (3 mM MgCl2, 400 µM of each dNTP and 50 U/mL Taq DNA polymerase; Promega, Madison, WI, USA). PCR conditions were: a partial denaturation at 95 °C for 10 min and 30 cycles with 45 sec at 95 °C, 30 sec at 52 °C−58 °C and 30 sec extension at 72 °C, and a final extension of 30 min at 72 °C was added in the last cycle. Fluorescently labeledCitation30 PCR products were separated on an Applied Biosystems 3730 capillary sequencer and scored using Gene Marker v1.95 (SoftGenetics LLC, State College, PA, USA).
Population genetic analysis
DNA fragment sizes, allele frequencies, and observed (Ho) and expected heterozygosity (He) assuming equilibrium conditions were determined using Cervus v3.0.3.Citation31 Missing data (no amplifications) were reported by loci but not considered in the analyses. We used a model-based clustering method for multilocus genotype data (13 microsatellites) to infer population structure and assign individuals to populations as implemented in Structure v2.3.3.Citation32,Citation33 The model assumes K populations (where K is inferred from the data) and each of which is characterized by a set of allele frequencies at each locus. Individuals are probabilistically assigned to populations, or jointly to two or more populations if their microsatellite data indicate that they are admixed. We evaluated the observed genetic diversity at different K values (K=2–10), and each K value was run independently 10 times with a burn-in period of 10 000 iterations followed by 10 000 iterations. Because individuals may have mixed ancestry, we applied the admixture model that assumes correlated allele frequency, in which each individual draws some fraction of his/her genome from each of the K populations. Structure Harvester Web v0.6.92Citation34 was used to compute Delta K values. R 2.15.1 (http://www.R-project.org) and ggplot2 (v0.9.1)Citation35 were used to graphically display the results.
Table 1 Each area, estimated population size and number of monkeys sampled by age class and sex
Testing for foamy virus antibodies
Western blot (WB)
All antibody testing was done using plasma samples. Rhesus macaque fibroblast Telo-RF (Tf) cell line was infected with SFV M. mulatta or M. fascicularisCitation21 or left uninfected. After 48 h, cells were lysed in 1× sodium disulfide sample buffer (12.5% 50 mM Tris-HCL (pH 6.8), 10% glycerol, 2% sodium disulfide, 5% 2-mercaptoethanol, 0.01% bromophenol blue) and samples electrophoresed through 10% polyacrylamide gels. After transfer of proteins to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA), the membranes were blocked in phosphate-buffered saline+10.05% Tween-20 (PBST) containing 2% nonfat dry milk, and rinsed with water and left to dry. Strips were made, each containing three lanes (molecular weight marker, uninfected Tf and infected Tf cell lysates) and primary antibody (monkey plasma) was added at a 1∶200 dilution and incubated overnight at 4 °C. Strips were washed three times in PBST before adding secondary goat anti-monkey horseradish peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at a 1∶5000 dilution for 1 h at room temperature (RT). Strips were washed three more times before rinsing with water and applying 3,3′,5,5′-tetramethylbenzidine stabilized substrate for horseradish peroxidase (Promega) to visualize bands.
Enzyme-linked immunosorbent assay (ELISA)
The general ELISA method has been described previously.Citation36 Ninety-six-well ELISA plates (Nalge Nunc, Rochester, NY, USA) were coated with 50 ng/well GST-Gag diluted in carbonate buffer (10 mM Na2CO3, 40 mM NaHCO3, pH 9.4) and incubated overnight at 4 °C. Heat-inactivated plasma were serially diluted from 1∶50 to 1∶1600 in blocking buffer and placed on the plates for 1 h at RT. Plates were washed three times with PBST. The plates were then incubated with a 1∶5000 dilution of goat anti-monkey horseradish peroxidase secondary antibody for 1 h at RT. Plates were washed three times with PBST and 100 µL 3,3′,5,5′-tetramethylbenzidine liquid substrate system (Sigma-Aldrich, St Louis, MO, USA) was added to each well and incubated at RT for 8 min. One hundred microliter 1 N H2SO4 was added to stop the reaction. Plates were read at 450 nm.
Testing for SRV-D antibodies
We and others have successfully used multiplex microbead immunoassays which require only a small volume (less than 5 μL) for the detection of antibodies to panels of viruses including SRV-D in nonhuman primates.Citation37,Citation38,Citation39 Using commercially available reagents from Charles River Laboratories (Wilmington, MA, USA), plasma samples were reacted with a liquid suspension array of microbeads coated with an SRV-D or other test or control antigens. After washing, the beads were reacted with biotinylated detector antibodies and then, after additional washing, Streptavidin-R-Phycoerthrin E. After a final washing to remove all unbound material, the beads were analyzed in a Bioplex reader (BioRad, Hercules, CA, USA). By monitoring the spectral properties of the beads and amount of Streptavidin-R-Phycoerthrin E fluorescence associated with each, the presence or absence of antibody to SRV-D antigens was determined.
FV PCR
DNA isolation from whole blood
Total genomic DNA was extracted from the whole blood of macaques using the QIAamp DNA blood mini kit (Qiagen, Valencia, CA USA) according to manufacturer’s protocol. Briefly, 200 µL of whole blood was mixed with proteases and equal volume of lysis buffer AL and 100% ethanol. The mixture was loaded onto columns and centrifuged. After two washes with AW1 and AW2 buffers, DNA was eluted twice with 200 µL of elution buffer AE.
PCR amplification of gag fragments from genomic DNA
A total of 10 µL of the isolated DNA was used for PCR amplification of gag fragments by a two round PCR. First round PCR with gag primers primer sequence: G1 (+) (nucleotide (nt) 114–134, taken from SFVmac 21010 sequence NC_001364 NC_001736) 5′-AGG ATGG TGG GGA CCA GCT A-3′; G2 (−) (nt 1451–1433) 5′-CAG GAA GAG GTA CTC GGG G-3′, was previously described.Citation40 The reactions were denatured at 95 °C for 3 min, followed by two cycles of 95 °C for 30 sec, 38 °C for 1 min, 72 °C for 1 min, then followed by 25 cycles of 95 °C for 30 sec, 52 °C for 1 min and 72 °C for 1 min. Water was used as a negative control. 2 µL of the first round PCR product was used as template for the second round PCR. We used gag primers G3 (+) (nt 190–212) 5′-CAA CCT AGA TGG AGA GCT GAA GG-3′; G4 (−) (nt 1425–1406) 5′-GGG GAC AAA CTA CGG CTG GG-3′, to amplify the gag fragment. The reactions were denatured at 95 °C for 3 min, followed by 35 cycles of 95 °C for 30 sec, 52 °C for 1 min and 72 °C for 1 min. The expected size of the PCR product for gag is 1235 bp.
Cloning and sequencing of SFV gag
The FV PCR products were gel purified using a QIAquick gel extraction kit (Qiagen) according to manufacturer’s protocol. The purified fragments were ligated into the TOPO-TA cloning vector (Invitrogen, Carlsbad, CA, USA) and transformed into Escherichi coli TOP10 competent cells (Invitrogen). Multiple colonies were individually selected from each gag sample and plasmid DNA was purified and sequenced. Sequencing was carried out using M13 forward and reverse primers, as well as internal forward and reverse primers for gag by GeneWiz Inc. (Seattle, WA, USA). SFV gag sequences generated in the current study have been deposited in GenBank with the following accession numbers KC991378 - KC992185.
SRV-D DNA testing
Real-time PCR was used to amplify the DNA extracted from whole blood to detect the presence of SRV-D (serotype 1) proviral DNA. SRV-D primers and probes were derived from highly conserved sequences within the envelope gene of SRV-D serotype 1, 2, 3, 4 and 5 viral genomes.Citation41 Primers: 5′-CTG GWC AGC CAA TGA CGG G-3′, 5'-CGC CTG TCT TAG GTT GGA GTG-3′; Probes: 6FAM—TCA CTA ACC TAA GAC AGG AGG GTC GTC A—TAMRA, 6FAM—TCC TAA ACC TAA GAC AGG AGG GCT GTC A—TAMRA. In addition, primers and probes designed to amplify the housekeeping gene oncostatin M were multiplexed into the reaction as a control for the presence of amplifiable DNA. 15 ul of isolated DNA was used in the following cycling conditions: 1 cycle for 2 min at 50 °C, 1 cycle for 10 min at 95 °C, 55 cycles for 15 s at 95 °C and 1 cycle for 1 min at 60 °C.
FV biocomputational analysis
Sequences were aligned using MUSCLE v3.8.31;Citation42 the relatively small number of apparent insertion/deletion events led to an alignment that was <0.3% gap. Amino-acid translation was performed using EMBOSS v6.4.0.0.Citation43
A large phylogenetic tree was built from the entire gag nucleic acid sequence alignment using FastTree v2.1.3 with a Jukes–Cantor model.Citation44 The tips of this tree were colored by sampling location using a python script together with the Bio.PhyloCitation45 BiopythonCitation46 library. This colored tree was rendered using Archaeopteryx v0.957 beta (http://www.phylosoft.org/archaeopteryx/). Additionally, a NeighborNet splits network was constructed using SplitsTreeCitation47 using LogDet distances and the NeighborNet algorithm.
Nucleic acid sequences were clustered using UCLUST v1.1Citation48 at a 97.5% identity, producing a total of 32 clusters. Three of these clusters (containing five sequences from animals MBG132, MBG189 and MBG110) clustered separately from other larger clusters as a result of G to A hypermutation (a more complete analysis in preparation for publication). When the putative hypermutation sites within these clusters were removed from the alignments, these sequences no longer clustered separately. Thus, these sequences were considered part of the clusters with which they clustered when these hypermutation sites were excluded; we define a viral ‘strain’ in the context of this study to be one of these clusters. For each macaque, the number of unique strains to which that animal’s sequences had been assigned was computed. Based on these counts, macaques were identified as either singly or multiply infected.
When the resulting clusters were ordered by size, there were six that were substantially larger than the rest. Each of these six was uniquely associated with a geographic region, except for Bormi, which had two clusters associated with it. We named each of these strains by the geographic location where it dominated; in the case of Bormi, the two clusters were arbitrarily designated Bormi1 and Bormi2. This assignment of viral strains is used in the rest of the present paper, furnishing the labeling in Figure 2.
An iterative method was employed to detect recombinant strains among these major viral strains. Two representative sequences were selected from each strain (except four for the Dhamrai strain) for recombination analysis with GARD,Citation49 using the HKY+Γ model. The trees generated from each non-recombinant alignment segment were inspected, and the strain with highest observed discord between trees was removed. This process was repeated until no recombination was detected within the alignment using GARD. This analysis removed both strains from the Bormi region, and left four strains found in the Charmaguria, Dhamrai, Dokhalo and Karamjal monkeys, which were identified as non-recombinant. Each step of the GARD results was confirmed by a PHI test analysisCitation50 as implemented in SplitsTree;Citation47 this test showed identical results with a P-value of less than 0.05 for each step. These four identified non-recombinant strains will be referred to as the ‘parental’ strains, which forms a subset of the above-defined ‘core’ strains.
The parental strains were used as genotype representatives for cBrother v2.0Citation51 analyses, where a representative from each cluster was evaluated for recombinant relationships to these potential parental strains. Two independent cBrother runs of 1.1 million generations were run for each cluster’s representative sequence, with the initial 10% discarded as burn-in, and sampling every 1000 generations. Convergence was assessed using the Gelman–Rubin diagnostic included with the cBrother distribution. Portions of evaluated sequences which were found to be descendant from a particular parental strain at 90% posterior probability or higher for at least 200 contiguous base pairs were identified as being partially descended from that parental strain.
For each of Bormi1 and Bormi2, which we had identified as recombinant strains in our GARD and PHI test analysis, two representative sequences were aligned with two representatives from each of the parental strains. These alignments were split at the corresponding breakpoints identified from the above cBrother analysis (304 and 496, respectively). Each of the resulting four alignments was analyzed using PhyML 3.0Citation52 under a HKY85 model with 100 bootstrap replicates and trees were generated using FigTree v1.3.1 (http://tree.bio.ed.ac.uk/software/figtree). The resulting trees put the recombinant sequences in the same positions as those inferred by cBrother.
Within-host pairwise distances were computed using R v2.15.1 (http://www.R-project.org) and the apeCitation53 package’s dist.dna function with the ‘raw’ model (normalized Hamming distance). Figure 3 was also created using R and ape.
Construction of a 6 nucleotide deletion mutant of gag in SFV-1
To create a gag gene containing the 6 nucleotide deletion in pSFV-1,Citation36 a two-step PCR protocol was used. The two primer sets used to PCR amplify overlapping fragments were: F1 (5′-GAG AGC TGA AGG TCG GGC TG-3′) R1 (5′-GGC CTG GTG ATG GTA AGG GTG AAG CCA-3′) and F2 (5′-GGC CTG GTG ATG GTA AGG GTG AAG CCA-3′) R2 (5′-CAC GAG CCT CTG ACT GCG GTT-3′). The outer primers F1 and R2 were then used to PCR amplify a 1.4 kb fragment (containing the deletion) using the overlapping fragments as template. The resulting PCR fragment and pSFV-1 vector were digested with BsaI and BsaBI, gel purified and ligated together. Several individual clones were sequenced to confirm the 6 nucleotide deletion.
Figure 2 Relationship between monkey and viral population structures. Each animal is represented by a single column spanning the figure’s sections. Columns are sorted by viral diversity, animal age, and sampling population. (A) Normalized Hamming distance for each pair of clones from each animal, representing intra-host viral diversity. The dotted line represents expected distance (due to PCR error) between distinct PCR products of a single DNA molecule. (B) The identifier of the animal to which each column corresponds. Respectively, (C), (D) and (E) represent number of viral clones, animal age class and sampling population for each monkey. (F) Microsatellite clusters as inferred by STRUCTURE; admixture between genotype clusters is represented by bar height. Microsatellite colors are assigned according to an observed correlation with sampling location. (G) Viral strains cloned from each animal, classified as described in the text. All sampled monkeys are represented except for one FV negative infant monkey (MBG211).

Assay of SFV-1 and deletion mutants
Transient transfection of Tf cells with pSFV-1 or the gag deletion mutant clones described above were performed following Qiagen’s Polyfect transfection protocol. Cells were harvested after 48 h and lysed for WB analysis, to detect Gag proteins. At the same time, extracellular supernatants from transfections were collected for an simian foamy-activated beta-glactosidase assay and clarified of cell debris by low-speed centrifugation.Citation21,Citation54 Simian foamy-activated beta-glactosidase cells are baby hamster kidney cells containing a β-galactosidase gene under the control of the SFV-1 long terminal repeat. When cells are infected with SFV, the viral tas gene activates the β-gal gene. To measure infectivity, a β-galactosidase expression assay was performed as previously described.Citation54
Results
M. mulatta populations used for sampling
Figure 1 is a map of Bangladesh with all the sampling sites indicated. A total of 164 rhesus macaques were sampled for the study, including 126 free ranging animals at six distinct sites, representing between 4% and 35% of the estimated population at each site (). Additionally, 38 performing macaques were sampled at five locations. Estimates of the total number of performing macaques in Bangladesh vary widely, from 500 to 5000. Because performing monkeys and their owners are nomadic and we have no a priori knowledge of the precise origins of the monkeys; for the purposes of our SFV gag diversity analyses we treat all 38 performing monkeys in this dataset as a single population.
Figure 3 Foamy virus sequence diversity overview. Phylogenetic tree built from all sequenced gag nucleotide clones using FastTree using the Jukes–Cantor sequence evolution model. Due to recombination, this tree should not be interpreted as an evolutionary history, but rather as an indication of the clustering seen in the sequence data. The edges of the tree are labeled with SH-like local supports.

Population structure of macaques using microsatellite DNA
Analysis of microsatellite DNA revealed five distinct genetic clusters, explained in part by geographic separation (Figures 2E and 2F). Supplementary Table S1 shows the number of alleles found for the 13 loci in different locations in Bangladesh and the observed heterozygosity (Ho) and expected heterozygosity (He) assuming equilibrium conditions. All the loci assayed were polymorphic with varying numbers of alleles (from 4 to 19, see Supplementary Table S1 for details). Out of the 13 loci, four were not in Hardy-Weinberg equilibrium. Using a Bayesian clustering algorithm,Citation55 the posterior probability for different numbers of clusters (K=2–10) was computed and the clustering patterns obtained with K=5 were associated with the highest delta K values (delta K=mean (|L″(K)|)/sd(L(K))) (structure harvester, available from: http://taylor0.biology.ucla.edu/struct_harvest/).Citation34
Each of the five microsatellite clusters identified in this analysis was found to be most closely associated with a particular macaque population, these being the Bormi, Charmaguria, Dhamrai, Karamjal and Narayanganj populations. Of these populations, Narayanganj stood out as having the greatest degree of admixture (i.e., combination of different lineages). The Dokhola monkeys seemed to be mostly admixtures of the Bormi and Karamjal genotypes. The reason for this apparent admixture is unclear. Most of the performing monkeys also bore a strong relationship to the Karamjal genotype. This relationship with Karamjal suggests a possible source for many of these performing monkeys.
Viral antibodies to FV Gag proteins
Initially we tested for FV antibodies using WB; 17 animals were tested in this way. The positive monkeys showed a clear doublet corresponding to the major gag proteins (data not shown). It became apparent that performing WB assays on all 164 animals sampled would not be feasible; therefore, we developed an ELISA assay using GST-Gag protein as the antigen. We performed ELISA on many of the WB-assayed animals and in all cases the WB and ELISA results matched. We therefore screened the remaining animals by ELISA. Negative animals showed no reactivity to GST-Gag at a 1∶50 plasma dilution, whereas positive animals showed clear reactivity at plasma dilutions of 1∶400 or greater (). We found that 149 of the 164 animals sampled have antibodies to SFV Gag. Details of the locations and ages of the FV antibody-positive and -negative animals are summarized in . SFV antibody-negative adult animals were only found in the performing monkey population. It is important to note that all the adults (n=89) in Dharmai, Bormi, Charmaguria, Narayanganj, Karamjal and Dokhola, were antibody-positive. Among these free-ranging populations, only 7 of the 36 younger animals, those estimated to be less than 24 months of age based on their dental eruption sequence, lacked SFV antibodies. Four out of five of the younger performing monkeys were SFV antibody positive as were 26/33 of the adult performing monkeys. Interestingly, all seven of the adult performing monkeys that were SFV negative were female.
Absence of SRV-D in sampled population
Asian macaques are the predominant natural hosts for SRV-D. Infections are not uncommon in captive macaque colonies, with prevalence varying widely from none to over 80%, determined at least partially by geographic origin and husbandry/management practices.Citation56 However, neither antibodies to SRV-D nor SRV-D viral DNA sequences were detected in any of the 164 animals sampled in the present study. While this finding is in sharp contrast to numerous reports of prevalence in captive colonies, there have been only limited reports of natural infections in the wild. These include less than 5% prevalence in Pongo pygmaeus (orangutan) from IndonesiaCitation57 and isolation of SRV-D of a novel type from Macaca mulatta and Semnopithecus entellus from Rajasthan, India.Citation58
The diagnostic antibody and PCR assays used in this study were developed and validated against the original known exogenous SRV-D 1, 2, 3, 4 and 5 serotypes.Citation39,Citation41 As reported previously, they are highly sensitive and specific and their cross-reactivity to more divergent serotypes that may be emerging is unknown. The SRV4 subtypes found in captive colony monkeys by investigators in Tsukuba, Japan and San Antonio, Texas were detected in the assays used for this study.Citation59,Citation60 However, other newer isolates such as SRV6 and SRV7 found in India have not been available for testing.Citation61
Table 2 Assay of anti-Gag antibodies using ELISA
Table 3 Geographic and age distribution of SFV seropositive rhesus macaques
Sequencing of SFV proviruses from peripheral blood
We initially screened antibody positive animals by PCR using a highly conserved portion of the integrase (IN) region of pol used by others for phylogenetic analyses.Citation62 However, for sequence analyses, we used primers to a portion of the gag gene which molecular genetics studies suggest would allow the most sequence diversity to be identified, since it only contains a small region with known function in viral replication.Citation63,Citation64,Citation65 We were able to amplify gag fragments from all of the SFV antibody-positive macaques. We obtained gag sequences from individual PCR products for each animal, with a median number of 6 gag clones per animal (Figure 2C).
Phylogenetic analyses of SFV gag
An initial phylogenetic analysis to gain an overview of SFV gag diversity in our samples (Figure 3) analysis showed that the viruses formed a number of tight clusters in the tree. As this initial analysis did not take viral recombination or other possible confounding effects into account, it only serves to visualize our concept of strains and suggests that there may be a correlation between viral genotype and sampling location. This observation was confirmed by our SplitsTreeCitation47 analysis (Figure 4). SplitsTree produces a ‘splits network’ capable of displaying non-treelike sequence difference information in a graphical form. In the figure, each set of parallel edges represents a ‘split’, which is a collection of sequence differences that separates one group of sequences from another. In this case, non-treelike evolution is expected to be from recombination; putative recombinant sequences are found at the end of the parallelograms formed by edges. This tree also shows clustering by region, and abundant recombinant sequences.
These observations were formalized by a clustering analysis using UCLUST v1.1Citation48 at a 97.5% identity threshold. The resulting clusters were used to define strains, as described in the section on ‘MATERIALS AND METHODS’. The six most populous strains were recognized as particularly representative of the bulk of the viral population diversity, and bore significant structural congruence with monkey population structure. These six strains will be called the ‘core’ strains and are noted as such in Figure 2G. Two of the strains, Bormi1 and Bormi2, were found to be recombinants of other core strains. The other, non-recombinant strains will be called ‘parental strains’. Differences between these strains are reflected on both nucleotide and amino-acid levels (Figure 5).
Figure 4 Splits network giving an overview of sequence diversity, and built by SplitsTree with strain labels. SplitsTree produces a ‘splits network’ capable of displaying non-treelike sequence difference information in a graphical form. In the figure, each set of parallel edges represents a ‘split’, which is a collection of sequence differences that separates one group of sequences from another. In this case, non-tree-like evolution is expected to be from recombination; putative recombinant sequences are found at the end of the parallelograms formed by edges. Strain labels have multiple identifiers if they were found to be recombinant by cBrother; e.g., a/b indicates a recombinant between core sequences a and b. An underscore (_) indicates the presence of a >200 bp region in which no one core strain was clearly found as the source.
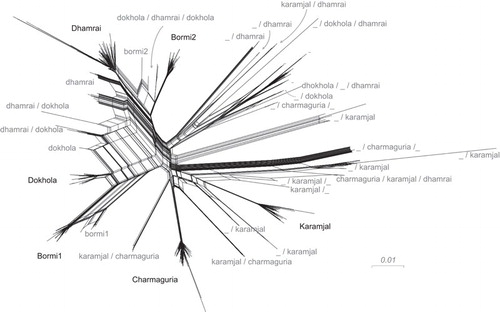
Of the monkey populations, three were strongly associated with a single core strain primarily found in that region: Charmaguria, Dhamrai and Karamjal. In these three areas, we only found the corresponding core strain, and not core strains from other areas. For convenience, we named these strains according to the sampling population with which they were closely associated.
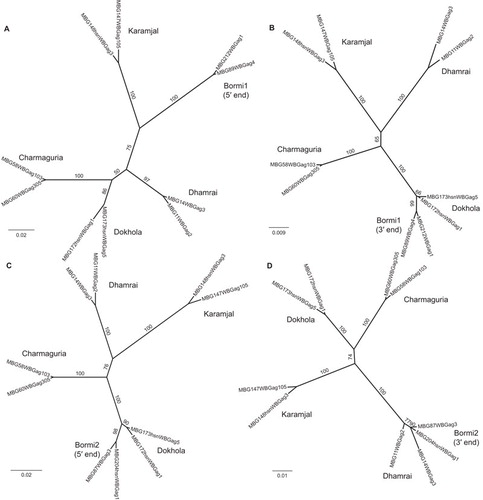
Two other populations, Bormi and Dokhola, stood out as having very structured viral populations. The Bormi monkey viruses separated into two distinct strains, almost entirely unique to this monkey population. We named these strains Bormi1 and Bormi2. These strains are not closely related (Figure 3). This separation of the virus population into two strains is not paralleled by a separation of the macaque population into two microsatellite groups (Figure 2F), suggesting two strains co-existing within a homogenous host population. The Dokhola monkey viruses also separated into two strains; one was identified as the Dhamrai strain, and another, seen predominantly in this population, was named the Dokhola strain. Recombination analysis using GARD,Citation49 cBrotherCitation51 and the PHI testCitation50 demonstrated that recombination played a role in shaping these viral populations. Charmaguria, Dokhola, Dhamrai and Karamjal core strains showed no evidence of recombination according to GARD or the PHI test. On the other hand, the two Bormi core strains each appeared to be derived from a recombinant lineage that has become established in the population. Bormi1 gag was found to be a recombinant form of some otherwise unsampled strain and the Dokhola strain, with a breakpoint at about nt 304 (Figures 6A and 6B). We were unable to identify a source for the first portion of this strain via a BLAST v2.2.25 search of either known Bangladeshi SFV viral strains or the NCBI nr (complete non-redundant) database, suggesting that this first portion is unique to the Bormi1 strain. The Bormi2 strain was found to be a recombinant form of the Dokhola and Dhamrai strains, with a breakpoint at about nt 496 in the 1125 nucleotide gag fragment we sequenced (Figures 6C and 6D).
The monkey populations with the least phylogenetically structured and most diverse viral populations were the Narayanganj and performing monkeys. Aside from a strong prevalence of the Dhamrai core strain within the Narayanganj monkeys, the viruses found in these populations were exceptionally divergent. Many of the very diverse viruses seen in the Narayanganj and performing populations were identified as recombinant in our cBrother analysis, while some appeared to be entirely distinct from the rest of the viral population. Additionally, all of the non-core strain viruses sampled from within Charmaguria, Dokhola and Karamjal were identified as having a recombinant relationship to the core strain associated with the sampled population in question.
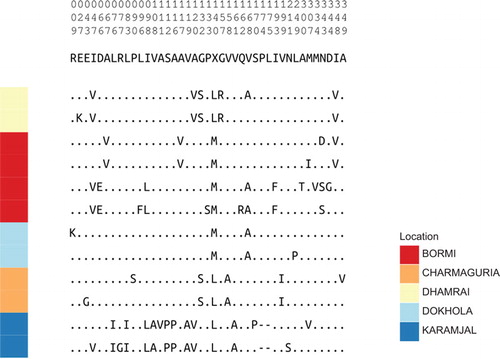
Figure 5 Amino-acid alignment of ‘Core’ strain representatives. Translated Gag sequence alignment between strain representatives compressed to variable sites. The numbers on the top of the figure show the position of these non-constant columns relative to the beginning of our gag sequencing region: the top digit is the 100’s place, the middle the 10’s place, and the bottom is the 1’s place.
We considered PCR-generated recombination unlikely given the very low copy number of FV in the samples (one copy per 104–106 cells); however, in order to eliminate the possibility that the observed recombinant sequences were generated during PCR, we did a simple DNA mixing experiment mimicking the PCR conditions used when processing samples. DNA preparations from two animals with single parental strains that were different from each other were mixed (e.g., MBG180 and MBG53). This was done with four different monkey pairs. After mixing the DNA preparations, PCR, cloning and sequence analyses were done using our standard conditions. Forty-five clones were analyzed (11–12 per DNA pair), and no recombinant sequences were detected, among these mixed templates. Only parental strains with minor PCR-generated errors were found. This is in contrast to the results for some macaques in which at least one recombinant was detected in six clones analyzed. This supports our conclusion that recombinants are present naturally and not generated during in vitro analysis. Additionally, the observation of recombinant strains seen at high frequency with the same break points (Bormi1 and Bormi2) further supports this position.
We would have liked to investigate co-evolution of SFV with the macaque host as in ref. 66; however, because we are looking at populations rather than a diverse collection of species, there was insufficient host viral genetic signal to confidently infer phylogenetic trees of the host. For this reason, we have instead used groupings based on population structure as described above.
SFV diversity within individual macaques
Variation in gag between clones isolated in individual macaques is indicative of more than one resident strain. A pairwise distance analysis was performed calculating the normalized number of differences between all pairs of gag sequences from a given monkey (Figures 2A and 2B). Rather than a continuum of distances, monkeys appeared to either have very low diversity, consistent with a single founding virus, or a large distance between some virus clones and a small distance between other pairs, consistent with two distinct infections. Although this observation could be explained via co-infection with two different viruses at a single time, we also noted a significant stratification of distances by monkey age. From this analysis, 76% of adult monkeys had single-virus infections, and 24% had multiple-virus infections, whereas 94% of juvenile monkeys had single-virus infections, and only 6% had multiple-virus infections (Figures 2A and 2D). A two-tailed Fisher’s exact test comparing the frequency of multiple-virus infections between age categories gave a P value of 0.009, indicating that there were significantly more multiple infections in older animals.
Amino acid analysis of the Gag protein
Since we obtained a considerable amount of gag nucleotide data, it was of interest to see how the Gag protein of the different strains varied at the predicted amino-acid level. The region of gag sequenced contains only one amino-acid motif known to be important for viral replication. This is the PSAP motif that is the late domain of Gag necessary for virus release.Citation63,Citation64 All the variants sequenced retain PSAP, even though the proviruses are latent. When we analyzed the putative amino-acid changes, the Bangladeshi viral strains are more similar to each than they are to an unrelated M. mulatta SFV1, which was isolated from a laboratory animal in the United States (). One change noted in the Karamjal strain is a 6 nucleotide (two amino acids) deletion. In order to assess whether this deletion has a functional consequence, it was created in the infectious clone pSFV1. We compared infectivity of SFV1 and SFV1 with the 6 nucleotide deletion (SFV1δ6nt) and found that there was little or no difference in replication competence in vitro (). Despite the fact that we are analyzing proviruses that are not expressed, we have no evidence for mutations that would compromise replication.
Figure 6 Recombination between Bormi strains and non-recombinant parental strains. Phylogenetic trees constructed by running PhyML on representative sequences which have been partitioned into separate alignments along recombination breakpoints inferred by cBrother. (A) and (B) show the relationship between Bormi1 and the core strains; (C) and (D) the relationship between Bormi2 and the core strains. (A) is built from sequence data between nucleotides 1 and 304, (B) from 305 on, (C) from 1 to 496 and (D) from 497 on (nucleotide (nt) number refers to position in the 1125 nt fragment of gag we sequenced, not to the nt position of the full length gag gene). Internal branches are labeled with their bootstrap values to indicate confidence.
Discussion
Synanthropy and disease risk
Rhesus macaques are the consummate synanthropic primate. Their ecological and behavioral flexibility allows them to thrive in human-altered environments. Among the Order Primates, only humans are more widely geographically distributed than the rhesus. Over centuries, facilitated by cultural traditions that encourage tolerance of NHP, rhesus macaques have come to inhabit niches in densely populated Bangladesh, where they come into frequent contact with humans. This long-term, complex interspecies association presents an opportunity to study how NHP-borne viruses originate, change and spread within and between NHP and human populations, and to better appreciate how the interplay between host ecology and viral evolution can potentially lead to the emergence of viruses of increased pathogenicity.
Foamy viruses naturally infect many mammalian species, including all NHP, cats and cows, as reviewed in ref. 14. Infants receive maternal antibodies against FV which protect them from infection. Young juveniles are not FV-infected. However, as NHP, cats and cows age, they become infected from older animals, which transmit the virus in saliva, through bites, licks or sharing of cud. Virtually, all adult animals that are not kept isolated from others of their species are infected. Infection is life-long, but there is no pathology associated with FV in natural hosts. DNA from latent proviruses is present in many tissues including peripheral blood, but virus and viral RNA indicative of replication is only detected in the oral mucosa and saliva. One study showed that in NHP, it is the superficial differentiating epithelial cells of the oral mucosal tissues that are infected.Citation17
Table 4 Percentage of amino-acid sequence similarityFootnotea in the region of gag analyzed between strains and SFV-1
Here we present results describing the prevalence and geographic distribution of the NHP-borne retrovirus, SFV, in Bangladeshi rhesus macaques that were sampled from multiple contexts. SFV infection was highly prevalent in free-ranging macaques, with all adult animals having been infected and 24% of the adults infected with more than one strain of SFV. This is consistent with rates of SFV infection measured in other free-ranging macaque populations;Citation21 however co-infection of single animals with more than one strain has not been well documented. Because our analyses only included 6–10 proviral clones per macaque, it is likely that the diversity we describe is an underestimate, and that we have missed some viral strains. Only 75% of the performing macaques we examined were infected by SFV. Performing macaques are often acquired by their owners as young animals, and subsequently, the animals have reduced contact with other NHP and hence, fewer opportunities to become infected with SFV.Citation67
Table 5 Infectivity of SFV-1 with a 6 nucleotide deletion in gag
Our analyses indicated that, for the majority of Bangladeshi macaques, SFV strains co-segregate with macaque populations, with one predominant strain associated with the majority of animals in a given area (Figure 2G). There were two exceptions to this finding. First, in Bormi, we found two distinct SFV core strains circulating in the macaque population, which we have called Bormi1 and Bormi2. Bormi samples from each of the 3 years under study contained both the Bormi1 and Bormi2 strains. Further longitudinal data are required to determine if these two strains will continue to persist or if one will eventually predominate in Bormi. Second, in the port city of Narayanganj, we observed a wide diversity of viruses. We found that several of these closely resembled viruses that predominate in other macaque populations, while others appeared to be novel. Additionally, several viruses from Narayanganj macaques appeared to be recombinants of core strains from other sites.
Biogeographic effects
Geographic factors may explain some of the observed patterns of microsatellite and SFV variation. Patterns of historical and contemporary forest cover and the country’s major rivers are likely to have influenced the geographic distribution of macaques and SFV. Like many countries in South and Southeast Asia, large areas of Bangladesh have been deforested over the past decades. As recently as 50 years ago, the forests of the Modhupur National Park covered much of central Bangladesh between the Padma, Jamuna and Meghna rivers. This extensive forest would have allowed the free movement of fauna, including subadult rhesus macaques, which normally leave their home groups and range in search of new groups. Phylogenetic analysis of SFV gag (Figure 3) indicated that the Bormi2 strain is a recombinant virus containing portions of the parental Dokhola and Dhamrai strains. Prior to widespread deforestation, Bormi, Dokhola and Dhamrai would all have fallen within the historical forests of the Modhupur Park (Figure 1), allowing macaques carrying the two parental strains to come into contact, setting the stage for viral recombination. In contrast, Charmaguria is located >30 km south of the Padma River and in a region of Bangladesh that has for decades lacked any significant, connected forest patches. The rhesus macaques in Charmaguria are effectively an isolated population. This isolation is reflected in the homogeneity seen in their microsatellites and SFV gag sequences.
Anthropogenic effects
Two groups stood out from the rest in terms of increased viral phylogenetic diversity: the performing macaques and the macaques from Narayanganj. Of the sites analyzed in this study, Narayanganj is associated with the highest human population density and the greatest commerce. It is located in the center of the country, close to the capital Dhaka and on a major river transit route. The diversity of viruses from the macaques in Narayanganj could be due to its use as a commercial hub. As with many Asian countries containing NHP habitats, pet NHP are common in Bangladesh and, as elsewhere, NHP owners face a dilemma when confronted with a monkey acquired as an infant that has grown into an aggressive adult. The solution is sometimes to release pets in an area with an existing NHP population.Citation68 It was not uncommon for us to find macaques with collars still about their necks in a free-ranging group. Narayanganj may simply be Bangladesh’s ‘melting pot’ for released and/or escaped pet macaques. Natural migration of macaques is unlikely to be a factor in Narayanganj, as there has been no forest within 50 km of Narayanganj for decades. Thus, the genetically and SFV heterogeneous monkey population that we observed in Narayanganj today is most likely a byproduct of human-assisted migration.
The observed genetic heterogeneity among the performing monkeys may be linked to the sociogeography and habits of nomadic people who travel throughout South Asia with their performing animals.Citation10,Citation69,Citation70 The Bade in Bangladesh have a centuries old tradition of capturing macaques from forested or urban areas and training them to perform for audiences. Performing monkeys are traded amongst nomadic groups, sometimes across national borders. Bade performing monkey owners stated that the Sundarban mangrove forest, near Karamjal, is a prime source for their monkeys, which is consistent with both our microsatellite and viral data.
Retroviral recombination
Our data show that individual animals can harbor more than one viral strain (Figures 2A and 2G). Whether infection with two viruses results from simultaneous infection from a single bite, transferring saliva containing multiple viruses, or from multiple bites is not known. However, our data showing a higher prevalence of multiple infections in older macaques are more consistent with the latter interpretation. This might imply that, SFV infection does not always protect from infection by other strains.
In this paper, we report the amplification and sequencing of DNA from blood samples and not from saliva. Analysis of SFV from the saliva of animals infected with multiple viral strains is needed to determine whether these animals also shed more than one viral strain, and this work is in progress. The existence of more than one viral strain in the monkey populations under study raises the possibility of historical and contemporary retroviral recombination. Indeed, our phylogenetic data suggest that recombination does occur in SFV. However, it is surprising that in Bormi, where two core strains are found, there is very little recombination detected so far between these strains. Further studies are needed to determine whether infection by a recombinant virus has biological consequences for the host.
Based on our results, we hypothesize that the observed patterns of genetic variation in SFV from these Bangladeshi rhesus macaques are explained, at least in part, by the human translocation of macaques. Translocation of infectious agents between populations of macaques sets the stage for simultaneous infection with more than one viral strain, as we detected in 17% of all macaques sampled regardless of age, and for the generation of recombinant viral genomes, which we also detected. Genetic recombination can produce retroviruses of altered phenotypes, as has been demonstrated for lentiviruses.Citation71 We have shown that frequent human/macaque contacts provide the contexts for NHP-to-human viral transmission,Citation26,Citation27 (companion manuscript in review). Future work will expand the scope and detail of our understanding of the interplay between host–pathogen–environment, as we are only beginning to appreciate how such complex dynamics at the human/NHP interface in Asia could affect the emergence of new diseases in humans.
Supplementary Figure 1
Download PDF (12.1 KB)The authors wish to thank our student assistants and faculty in the Wildlife Branch of the Department of Zoology at Jahangirnagar University, Bangladesh. We also thank the Bangladesh Forest Department for their permission and constant support of our research program. Drs John Heidrich (Ventana Animal Clinic, Albuquerque, NM, USA) and Michael Schillaci (University of Toronto Scarborough, Toronto, Ont., Canada) as well as Hanna and Leah Engel (Seattle, WA, USA) and Lynn Johnson (National Geographic Magazine, Washington, DC, USA) were outstanding assistants during multiple field seasons. We thank Robin Watanabe (University of Washington, Seattle, WA, USA), Ann Rosenthal, Shannon Flynn (University of California, Davis, CA, USA), Connor McCoy, Leah Engel and Brandon Duffy (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) for their laboratory technical assistance and discussions and Amanda Zeller and Regina Liszanckie (University of Washington, Seattle, WA, USA) for administrative support. We are grateful to Drs Harmit Malik and Michael Emerman (Fred Hutchinson Cancer Research Center, Seattle, WA, USA) for their insightful suggestions and comments. This research was supported by funding from NIH-NIAID grants R01 AI078229, R01AI078229-03S1, R03 AI064865, NIH-NCI grant CA18282, NIH-NCRR grant P51 RR000166 and New Development Institutional Support from the Fred Hutchinson Cancer Research Center. Supplementary Information accompanies the paper on Emerging Microbes & Infections's website (http://www.nature.com/EMI/).
Supplementary information accompanies the paper on Emerging Microbes & Infections's website (http://www.nature.com/EMI/).
- Bailes E, Gao F, Bibollet-Ruche F et al.Hybrid origin of SIV in chimpanzees. Science2003;300: 1713–1716.
- Daszak P, Cunningham AA, Hyatt AD.Anthropogenic environmental change and the emergence of infectious diseases in wildlife. Acta Trop2001;78: 103–116.
- Jones KE, Patel NG, Levy MA et al.Global trends in emerging infectious diseases. Nature2008;451: 990–993.
- Weiss RA, McMichael AJ.Social and environmental risk factors in the emergence of infectious diseases. Nat Med2004;10 (12 Suppl): S70–S76.
- Plowright RK, Foley P, Field HE et al.Urban habituation, ecological connectivity and epidemic dampening: the emergence of Hendra virus from flying foxes (Pteropus spp.). Proc Biol Sci2011;278: 3703–3712.
- Engering A, Hogerwerf L, Slingenbergh J.Pathogen–host–environment interplay and disease emergence. Emerg Microbes Infect2013;2: e5.
- Khatun UH,Ahsan MF,Røskaft E.Attitudes of the local community towards the conservation of the common langur (Semnopithecus entellus) in Keshabpur, Bangladesh. Int J Biodivers Conserv2012;4: 385–399.
- Feeroz MM.Species diversity and population density of non-human primates in north-east and south-east of Bangladesh. Ecoprint2011;8: 53–57.
- Hasan MK, Feeroz MM.Distribution and status of long-tailed macaque (Macaca fascicularis aurea I. Geoffroy Saint-Hilaire, 1830) in Bangladesh. J Threat Taxa2010;2: 1342–1344.
- Mack TM, Noble J Jr.Natural transmission of smallpox from man to performing monkeys. An ecological curiosity. Lancet1970;1: 752–754.
- Oberste MS, Feeroz MM, Maher K et al.Characterizing the picornavirus landscape among synanthropic nonhuman primates in Bangladesh, 2007 to 2008. J Virol2013;87: 558–571.
- Oberste MS, Feeroz MM, Maher K et al.Naturally acquired picornavirus infections in nonhuman primates at the Dhaka Zoo. J Virol2013;87: 572–580.
- Karlsson EA, Engel GA, Feeroz MM et al.Influenza virus infection in nonhuman primates. Emerg Infect Dis2012;18: 1672–1675.
- Linial ML.Foamy viruses.In: Knipe DM, Howley PM (ed.) Fields virology.5th ed.Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins, 2007: 2245–2263.
- Han GZ, Worobey M.An endogenous foamy-like viral element in the coelacanth genome. PLoS Pathog2012;8: e1002790.
- Linial ML.Why aren’t foamy viruses pathogenic? Trends Microbiol2000;8: 284–289.
- Murray SM, Picker LJ, Axthelm MK, Hudkins K, Alpers CE, Linial ML.Replication in a superficial epithelial cell niche explains the lack of pathogenicity of primate foamy virus infections. J Virol2008;82: 5981–5985.
- Falcone V, Leupold J, Clotten J et al.Sites of simian foamy virus persistence in naturally infected African green monkeys: latent provirus ubiquitous, whereas viral replication is restricted to the oral mucosa. Virology1999;257: 7–14.
- Voevodin AF, Marx PA.Spumaviruses.In: Simian virology.London: Blackwell Publishing Ltd, 2009: 217–236.
- Lerche NW.Simian retroviruses: infection and disease—implications for immunotoxicology research in primates. J Immunotoxicol2010;7: 93–101.
- Jones-Engel L, Steinkraus KA, Murray SM et al.Sensitive assays for simian foamy viruses reveal a high prevalence of infection in commensal, free-ranging, Asian monkeys. J Virol2007;81: 7330–7337.
- Boneva RS, Switzer WM, Spira TJ et al.Clinical and virological characterization of persistent human infection with simian foamy viruses. AIDS Res Hum Retroviruses2007;23: 1330–1337.
- Switzer WM, Bhullar V, Shanmugam V et al.Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J Virol2004;78: 2780–2789.
- Switzer WM, Garcia AD, Yang C et al.Coinfection with HIV-1 and simian foamy virus in west Central Africans. J Infect Dis2008;197: 1389–1393.
- Sandstrom PA, Phan KO, Switzer WM et al.Simian foamy virus infection among zoo keepers. Lancet2000;355: 551–552.
- Jones-Engel L, Engel GA, Schillaci MA et al.Primate-to-human retroviral transmission in Asia. Emerg Infect Dis2005;11: 1028–1035.
- Jones-Engel L, May CC, Engel GA et al.Diverse contexts of zoonotic transmission of simian foamy viruses in Asia. Emerg Infect Dis2008;14: 1200–1208.
- Hasan MK, Aziz MA, Alam SMR et al.Distribution of rhesus macaques (Macaca mulatta) in Bangladesh: inter-population variation in group size and composition. Primate Conserv2013;26: 125–132.
- Jones-Engel L, Engel GA, Heidrich J et al.Temple monkeys and health implications of commensalism, Kathmandu, Nepal. Emerg Infect Dis2006;12: 900–906.
- Kanthaswamy S, von Dollen A, Kurushima JD et al.Microsatellite markers for standardized genetic management of captive colonies of rhesus macaques (Macaca mulatta). Am J Primatol2006;68: 73–95.
- Kalinowski ST, Taper ML, Marshall TC.Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol Ecol2007;16: 1099–106.
- Pritchard JK, Stephens M, Donnelly P.Inference of population structure using multilocus genotype data. Genetics2000;155: 945–959.
- Falush D, Stephens M, Pritchard JK.Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics2003;164: 1567–1587.
- Earl DA, vonHoldt BM.STRUCTURE HARVESTER: a website and program for visualizing structure output and implementing the Evanno method. Conservation Genetics Resources2012;4: 359–361.
- Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer, 2009.
- Mergia A, Wu M.Characterization of provirus clones of simian foamy virus type 1. J Virol1998;72: 817–822.
- Kuller L, Watanabe R, Anderson D, Grant R.Development of a whole-virus multiplex flow cytometric assay for antibody screening of a specific pathogen-free primate colony. Diagn Micr Infec Dis2005;53: 185–193.
- Khan IH, Mendoza S, Yee J et al.Simultaneous detection of antibodies to six nonhuman-primate viruses by multiplex microbead immunoassay. Clin Vaccine Immunol2006;13: 45–52.
- Liao Q, Guo H, Tang M et al.Simultaneous detection of antibodies to five simian viruses in nonhuman primates using recombinant viral protein based multiplex microbead immunoassays. J Virol Methods2011;178: 143–152.
- Schweizer M, Neumann-Haefelin D.Phylogenetic analysis of primate foamy viruses by comparison of pol sequences. Virology1995;207: 577–582.
- White JA, Todd PA, Rosenthal AN et al.Development of a generic real-time PCR assay for simultaneous detection of proviral DNA of simian Betaretrovirus serotypes 1, 2, 3, 4 and 5 and secondary uniplex assays for specific serotype identification. J Virol Methods2009;162: 148–154.
- Edgar RC.MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics2004;5: 113.
- Rice P LIBA.EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet2000;16: 276–277.
- Price MN, Dehal PS, Arkin AP.FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol2009;26: 1641–1650.
- Talevich E, Invergo BM, Cock PJ, Chapman BA.Bio.Phylo: a unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinformatics2012;13: 209.
- Cock PJ, Antao T, Chang JT et al.Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics2009;25: 1422–1423.
- Huson DH, Bryant D.Application of phylogenetic networks in evolutionary studies. Mol Biol Evol2006;23: 254–267.
- Edgar RC.Search and clustering orders of magnitude faster than BLAST. Bioinformatics2010;26: 2460–2461.
- Kosakovsky Pond SL, Posada D, Gravenor MB, Woelk CH, Frost SD.GARD: a genetic algorithm for recombination detection. Bioinformatics2006;22: 3096–3098.
- Bruen TC, Philippe H, Bryant D.A simple and robust statistical test for detecting the presence of recombination. Genetics2006;172: 2665–2681.
- Fang F, Ding J, Minin VN, Suchard MA, Dorman KS.cBrother: relaxing parental tree assumptions for Bayesian recombination detection. Bioinformatics2007;23: 507–508.
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O.New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biol2010;59: 307–321.
- Paradis E, Claude J, Strimmer K.APE: analyses of phylogenetics and evolution in R language. Bioinformatics2004;20: 289–290.
- Yu SF, Linial ML.Analysis of the role of the bel and bet open reading frames of human foamy virus by using a new quantitative assay. J Virol1993;67: 6618–6624.
- Hubisz MJ, Falush D, Stephens M, Pritchard JK.Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour2009;9: 1322–1332.
- Nandi JS, Bhavalkar-Potdar V, Tikute S, Raut CG.A novel type D simian retrovirus naturally infecting the Indian Hanuman langur (Semnopithecus entellus). Virology2000;277: 6–13.
- Warren KS, Niphuis H, Heriyanto, Verschoor EJ, Swan RA, Heeney JL.Seroprevalence of specific viral infections in confiscated orangutans (Pongo pygmaeus). J Med Primatol1998;27: 33–37.
- Nandi JS, Van DS, Chhangani AK, Mohnot SM.New simian beta retroviruses from rhesus monkeys (Macaca mulatta) and langurs (Semnopithecus entellus) from Rajasthan, India. Virus Genes2006;33: 107–116.
- Zao CL, Armstrong K, Tomanek L et al.The complete genome and genetic characteristics of SRV-4 isolated from cynomolgus monkeys (Macaca fascicularis). Virology2010;405: 390–396.
- Hara M, Sata T, Kikuchi T et al.Isolation and characterization of a new simian retrovirus type D subtype from monkeys at the Tsukuba Primate Center, Japan. Microbes Infect2005;7: 126–131.
- Nandi JS, Tikute SA, Chhangani AK et al.Natural infection by simian retrovirus-6 (SRV-6) in Hanuman langurs (Semnopithecus entellus) from two different geographical regions of India. Virology2003;311: 192–201.
- Hussain AI, Shanmugam V, Bhullar VB et al.Screening for simian foamy virus infection by using a combined antigen Western blot assay: evidence for a wide distribution among Old World primates and identification of four new divergent viruses. Virology2003;309: 248–257.
- Stange A, Mannigel I, Peters K et al.Characterization of prototype foamy virus gag late assembly domain motifs and their role in particle egress and infectivity. J Virol2005;79: 5466–5476.
- Patton GS, Morris SA, Chung W, Bieniasz PD, McClure MO.Identification of domains in gag important for prototypic foamy virus egress. J Virol2005;79: 6392–6399.
- Rethwilm A.Molecular biology of foamy viruses. Med Microbiol Immunol2010;199: 197–207.
- Switzer WM, Salemi M, Shanmugam V et al.Ancient co-speciation of simian foamy viruses and primates. Nature2005;434: 376–380.
- Blewett EL, Black DH, Lerche NW, White G, Eberle R.Simian foamy virus infections in a baboon breeding colony. Virology2000;278: 183–193.
- Jones-Engel L, Engel GA, Schillaci MA, Babo R, Froehlich J.Detection of antibodies to selected human pathogens among wild and pet macaques (Macaca tonkeana) in Sulawesi, Indonesia. Am J Primatol2001;54: 171–178.
- Schillaci MA, Jones-Engel L, Engel GA et al.Prevalence of enzootic simian viruses among urban performance monkeys in Indonesia. Trop Med Int Health2005;10: 1305–1314.
- Berland JC. No five fingers are alike.Cambridge, MA: Harvard University Press, 1982.
- Gao F, Bailes E, Robertson DL et al.Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature1999;397: 436–441.