
Abstract
Highly pathogenic H5N1 and low pathogenic H9N2 influenza viruses are endemic to poultry markets in Bangladesh and have cocirculated since 2008. H9N2 influenza viruses circulated constantly in the poultry markets, whereas highly pathogenic H5N1 viruses occurred sporadically, with peaks of activity in cooler months. Thirty highly pathogenic H5N1 influenza viruses isolated from poultry were characterized by antigenic, molecular, and phylogenetic analyses. Highly pathogenic H5N1 influenza viruses from clades 2.2.2 and 2.3.2.1 were isolated from live bird markets only. Phylogenetic analysis of the 30 H5N1 isolates revealed multiple introductions of H5N1 influenza viruses in Bangladesh. There was no reassortment between the local H9N2 influenza viruses and H5N1 genotype, despite their prolonged cocirculation. However, we detected two reassortant H5N1 viruses, carrying the M gene from the Chinese H9N2 lineage, which briefly circulated in the Bangladesh poultry markets and then disappeared. On the other hand, interclade reassortment occurred within H5N1 lineages and played a role in the genesis of the currently dominant H5N1 viruses in Bangladesh. Few ‘human-like’ mutations in H5N1 may account for the limited number of human cases. Antigenically, clade 2.3.2.1 H5N1 viruses in Bangladesh have evolved since their introduction and are currently mainly homogenous, and show evidence of recent antigenic drift. Although reassortants containing H9N2 genes were detected in live poultry markets in Bangladesh, these reassortants failed to supplant the dominant H5N1 lineage.
Introduction
The highly pathogenic avian influenza (HPAI) A (H5N1) virus first appeared in 1996 in geese in Guangdong, China, and continues to circulate in the poultry population in many countries, mainly in Asia. Although the HPAI H5N1 virus has not yet demonstrated the ability to transmit efficiently between humans, it remains a public health concern because of the high case-fatality rates associated with H5N1 infection. Recent studies on ferrets show that limited changes are required for the H5N1 virus to acquire the ability for airborne transmission.Citation1,Citation2
Of the 32 distinguishable genetic clades of H5N1 viruses,Citation3 those that are currently circulating are clades 1.1 (Cambodia and Vietnam); 2.1.3.2 (Indonesia); 2.2.1 (Egypt); 2.3.2.1 (Bangladesh, China, India, Indonesia, Korea, Nepal and Vietnam); 2.3.4.2 (China); and 7.2 (China and Vietnam).Citation4 Clade 2.3.2.1 has been particularly associated with wild-bird infections,Citation3 and after it was first detected in migratory birds in Mongolia in 2009,Citation5 it spread to eastern Europe in 2010.Citation6,Citation7
Bangladesh has one of the highest densities of human (1072 people/km2) and poultry populations (1194 birds/km2) in the world.Citation8 Approximately 75% of the human population in Bangladesh depends heavily on poultry as a source of meat and household income.Citation8 Thus, there is a high risk of rapid disease spread and transmission to a large number of people in the event that a virus with a pandemic potential emerges in Bangladesh.
The HPAI H5N1 virus was first detected in Bangladesh on 5 February 2007, on a poultry farm in Jamalpur district, Dhaka division, and as of 1 November 2013, there have been 549 outbreaks reported in the country.Citation9 From February 2007 until the end of 2010, the circulating HPAI H5N1 viruses in Bangladesh were all from clade 2.2, sublineage III; however, at the beginning of 2011, new introductions of clades 2.3.2.1 and 2.3.4.2 were detected.Citation10,Citation11
As of 1 November 2013, seven human cases of HPAI H5N1 infection have been reported in Bangladesh.Citation12 All the cases had a history of exposure to poultry, including sick or dead birds. The first three cases occurred in 2008–2011 in Kamalpur, Dhaka, in children under the age of two years, and the viruses isolated from the first and the second case belonged to clades 2.2 and 2.2.2, respectively.Citation4,Citation13 Three cases were reported in 2012 in adult males working at the live bird markets in Dhaka city and all three viruses belonged to clade 2.3.2.1.Citation4 On 18 February 2013, the seventh case and first human death caused by HPAI H5N1 was reported in a 23-month-old boy from Comilla. Influenza A (H5N1) virus from clade 2.3.2.1 was isolated from the patient’s respiratory swab and from tissue samples from sick chickens with which the boy had been in contact.Citation14
In addition to the sporadic spread of H5N1 viruses, H9N2 influenza viruses have been isolated year-round from birds at the live bird markets in Dhaka city since 2008.Citation15,Citation16 Before these studies, isolation of H9N2 viruses in Bangladesh had not been reported. However, on the basis of molecular and phylogenetic analyses, Shanmuganatham et al.Citation16 hypothesized that the H9N2 subtype had been introduced into Bangladesh from Pakistan through poultry movements in the early 2000s.
In this study, we performed antigenic, molecular and phylogenetic characterization of the dominant H5N1 lineages in Bangladesh from November 2009 to June 2013 and found high homology among the majority of H5N1 viruses. We also detected the separate introduction of two reassortant HPAI viruses of subtype H5N1 as well as interclade reassortment, but did not find reassortants with cocirculating H9N2 viruses.
MATERIALS AND METHODS
Sample collection
This study report results from ongoing active avian influenza surveillance in Bangladesh that started in November 2008. The target locations for sample collection were five retail poultry markets and a pet bird market in Dhaka city; six poultry farms (chicken and duck farms) in the Dhaka and Rajsashi divisions; the Baikka Beel wetlands in Sreemangal, located in the Moulvibazar district (200 km northeast of Dhaka); and a lake in Savar, Dhaka, which serves as a feeding station for migratory wild birds. Samples from the retail markets and the farms were collected throughout the year, whereas environmental samples from the wetlands and the lake were collected only during the winter and spring seasons when the wild birds are present during migration.
Sample screening and virus isolation
All samples were screened for avian influenza viruses (AIVs) by real-time reverse transcription polymerase chain reaction, using influenza A-specific primers and probesCitation17 as previously described.Citation15 All influenza A real-time reverse transcription polymerase chain reaction-positive samples were inoculated into 10-day-old embryonated chicken eggs to confirm the presence or absence of virus infection.
Viral RNA was extracted from allantoic fluid, using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and isolates were initially screened by sequencing for influenza viruses subtypes H5 and H9 and the Newcastle disease virus, since all three viruses commonly circulate in Bangladesh.
Antigenic analysis
Preparation of post-infection ferret antisera for hemagglutination inhibition (HI) assays
All post-infection ferret antisera used in the HI assays in this study were produced in United States Department of Agriculture-approved biosafety level 3 enhanced facilities at St Jude Children’s Research Hospital (St Jude, Memphis, TN, USA). Because the antisera α-RG-A/barn swallow/Hong Kong/1161/2010 (H5N1), α-A/environment/Bangladesh/15121/2012 (H5N1) and α-A/duck/Bangladesh/19097/2013 (H5N1) induced very low antibody titers after infection, the immune response in ferrets was boosted with Freund’s incomplete adjuvant (Invivogen, Inc., San Diego, CA, USA) at day 14 post-infection. Initially, each of three groups of three ferrets was infected intranasally with one of the following live viruses: 0.5 mL 107 50% egg infectious dose (EID50) of wild-type A/duck/Bangladesh/19097/2013(H5N1), 0.5 mL 107 EID50 of wild-type A/environment/Bangladesh/15121/2012 (H5N1) or 1.0 mL 108 EID50 of reverse genetics 2+6 A/barn swallow/Hong Kong/1161/2010 (H5N1), A/Puerto Rico/8/34 (internal genes). Fourteen days post-infection, the surviving ferrets were bled and then boosted intramuscularly with 0.5 mL 109 EID50 of the homologous virus, with Freund’s incomplete adjuvant according to the instructions of the manufacturer. One ferret from the group receiving A/environment/Bangladesh/15121/2012 (H5N1) did not survive the initial infection.
HI assays
HI assays were done as previously described.Citation18 The panel of antisera used in the HI assays included representatives from the currently circulating genetic lineages of clade 2.3.2.1 in Asia and Bangladesh, as well as a representative from clade 2.2. Horse red blood cells (1%) were used for the assays.
Sequencing and phylogenetic analysis
DNA sequencing was performed by the Hartwell Center for Bioinformatics and Biotechnology at St Jude, using universal primersCitation19 and subtype-specific primers (primer sequences available upon request), and the BigDye Terminator v3.1 Cycle Sequencing Kit (Invitrogen, Carlsbad, CA, USA) on 3730XL DNA analyzers (Applied Biosystems, Foster City, CA, USA), according to the manufacturer’s recommendations.
The hemagglutinin (HA) and neuraminidase (NA) genes of all 30 H5N1 isolates from Bangladesh were sequenced, and 23 isolates from 2010 to 2013 were selected for complete genome sequencing (). Full-genome phylogenetic analysis of the sequenced Bangladeshi H5N1 viruses was performed, and representative nucleotide (nt) sequences of H5N1 clades that have been recently circulating in Asia and Egypt were retrieved from the National Center for Biotechnology Information (NCBI) Influenza Virus Sequence DatabaseCitation20 and the EpiFlu database of the Global Initiative on Sharing All Influenza DataCitation21. All nucleotide sequences were aligned by using the ClustalW tool in MEGA 5 and trimmed to equal lengths, but at least 90% of the coding region as follows: HA, 1630 nt; NA, 1350 nt; polymerase basic protein 1 (PB1), 2213 nt; polymerase basic protein 2(PB2), 2214 nt; polymerase acidic (PA), 2069 nt; nucleoprotein (NP), 1416 nt; Matrix (M), 744 nt; and non-structural (NS), 693 nt. Phylogenetic relationships were inferred by the neighbor-joining method from 1000 bootstrap values, the topology was confirmed by the maximum likelihood method, and evolutionary analyses were conducted in MEGA 5.Citation22
Table 1 Avian influenza A (H5N1) virus isolates from live bird markets in Bangladesh from December 2010 to March 2013
The sequences obtained in this study were deposited in the Influenza Research Database and are available under GenBank accession numbers: AGZ62382, AGZ62383, KF874284–KF874289 and KF888395–KF888584.
RESULTS
Influenza A viruses in Bangladeshi live poultry markets
From November 2008 to June 2013, 975 low pathogenic avian influenza (LPAI) H9N2 viruses and 66 HPAI H5N1 viruses were isolated from 19 897 biological and environmental samples collected from apparently healthy birds in Bangladesh. Co-infection with both HPAI H5N1 and LPAI H9N2 viruses was found in 78 samples from single hosts (Figure ). Influenza viruses from subtypes other than H5 and H9 were isolated very rarely, mainly H3N8 in one of the duck farms in 2012.
HPAI H5N1 viruses were isolated only from the live bird markets from three bird species: chickens, quail and ducks. AIVs were not detected in samples from migratory wild waterfowl collected at the wetland and the lake.
Figure 1 Samples collected and avian influenza viruses isolated in Bangladesh (November 2008–June 2013). Total number of samples collected was 19 897 (3952 oropharyngeal, 4113 cloacal and 11 832 environmental samples, the last set consisting of 1044 water and 10 788 fecal samples). The number of samples collected by bird species is as follows: chickens, 11 513; ducks, 1616; quail, 1460; pet birds, 2350; lesser whistling ducks, 2196; and unidentified wild waterfowl, 725.
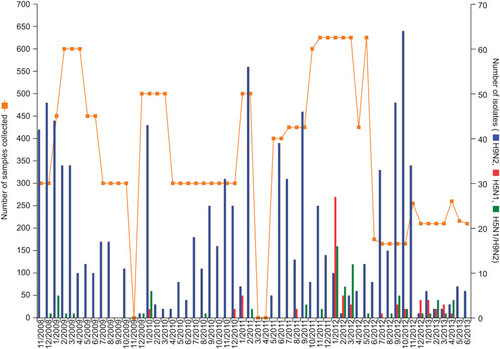
During the study period, H9N2 viruses were isolated every month except September 2009 and there was no specific pattern of seasonality in virus prevalence. H5N1 viruses were mainly isolated during the winter and spring seasons of each year, but sporadically one to five H5N1 viruses or a co-infection with H5N1 and H9N2 in a single-host sample were detected from August to October. Figure shows the number of samples collected and viruses isolated every month during the surveillance period.
Antigenic analysis in HI assays
presents results of the HI assays with 30 H5N1 isolates from Bangladesh, using the World Health Organization reference antisera against H5N1 vaccine candidate strains from clades 2.3.2.1 and 2.2 and ferret sera produced against the H5N1 viruses isolated from Bangladesh.
Table 2 Antigenic analysis of H5N1 influenza viruses from Bangladesh in the hemagglutination inhibition assays
The two H5N1 isolates from clade 2.2.2 (A/Ck/BD/9636/10 (H5N1) and A/Ck/BD/9675/11 (H5N1)) cross-reacted with antisera to clades 2.2.2 and 2.3.2.1 viruses in the HI assays. Their HI titers against α-A/Bar-headed goose/Qinghai/1A/05 (H5N1), clade 2.2, were within 1 log2 difference from the homologous titer of that serum, and were as high as the HI titers that the clade 2.2.2 isolates showed against the sera produced to the recent Bangladeshi clade 2.3.2.1 isolates α-A/Ck/BD/15205/12 (H5N1) and α-A/Env/BD/15121/12 (H5N1). In contrast, the α-A/Bar-headed goose/Qinghai/1A/05 (H5N1), clade 2.2, antiserum cross-reacted with clade 2.3.2.1 viruses with very low HI titers (≤40).
Clade 2.3.2.1 viruses from Bangladesh reacted against all clade 2.3.2.1 antisera. The cross-reactivity was lowest to α-A/Hubei/1/10 (H5N1) (clade 2.3.2.1), with HI titers between 10 and 160, whereas the homologous titer of the antiserum was 640. The clade 2.3.2.1 viruses had highest HI titers against α-A/Env/BD/15121/12 (H5N1) (clade 2.3.2.1) and α-A/Barn swallow/HK/1161/10 (H5N1) (clade 2.3.2.1), followed by α-A/Dk/BD/19097/13 (H5N1) (clade 2.3.2.1). The high HI titers obtained with these sera (1/2560–1/5120) were because the immune response was boosted with an adjuvant, but this would likely not affect the antigenicity pattern of the viruses. The HI titers of the clade 2.3.2.1 isolates against α-A/Ck/BD/15205/12 (H5N1) (clade 2.3.2.1) were within 1 log2 difference of its homologous titer. This observation confirmed that sera specifically raised to recent Bangladeshi isolates provided the highest level of HI. Clade 2.3.2.1 viruses from Bangladesh also reacted well to α-A/Common magpie/HK/5052/07 (H5N1) (clade 2.3.2.1), but the HI titers were up to 4 logs2 lower than the homologous titer. Three recent isolates from clade 2.3.2.1—A/Quail/BD/19250/13 (H5N1), A/Quail/BD/19254/13 (H5N1) and A/Env/BD/19338/13 (H5N1)—had very low HI titers (≤40 with all non-boosted sera and 160–640 with boosted sera), supporting the occurrence of antigenic drift among recent H5N1 isolates.
Molecular characteristics of Bangladeshi H5N1 viruses
Alignment of the sequences of the H5N1 viruses isolated in this study with representative sequences from NCBI and Global Initiative on Sharing All Influenza Data identified some amino-acid residues in each genomic segment that were molecular signatures of the viruses isolated in the South Asian geographic region encompassing Bangladesh–India–Nepal (). This molecular signature was clade-specific.
Table 3 Amino-acid residues characteristic of the H5N1 viruses isolated from Bangladesh
HA gene
All H5N1 viruses isolated in Bangladesh had multiple basic amino acids at the cleavage site (CS) at positions 321–333 of the HA (H5 numbering), which is a marker for high pathogenicity in chickens.Citation26 Clade 2.2.2 isolates had a PQGERRRKKR/GLF CS sequence, which was identical with the HA CS motif of wild-bird H5N1 isolates from Qinghai Lake in 2005Citation27 and clade 2.2.2 isolates from the India and Bangladesh geographic region, including the human isolate A/BD/3233/2011 (H5N1) (GenBank accession number AEA50985).
Of the 28 clade 2.3.2.1 isolates, 26 had the PQRERRRKR/GLF CS motif, including deletion at position 329, which is common in clade 2.3.2.1 H5N1 viruses from the Bangladesh, India and Nepal regions and from wild birds from Qinghai (China), Mongolia, and Tyva in 2009 and 2010. Two isolates from clade 2.3.2.1 had an amino-acid variation at position 323 of the CS: A/Ck/BD/15210/12 (H5N1) had R323G and A/Env/BD/19338/13 (H5N1) had R323K.
lists the amino-acid residue positions at the HA antigenic sites of isolates from clades 2.2.2 and 2.3.2.1. Residue R189G found in several clade 2.3.2.1 isolates from Bangladesh was unique, whereas the others residues were common in H5N1 viruses from Bangladesh, India and Nepal. HA residues 94N, 155N, 156A, 189R and 226I, which we found to occur in the majority of H5N1 viruses isolated in Bangladesh, have been reported to increase virus binding to α-2,6 sialic acid receptors ().Citation23,Citation24,Citation25 Residues 222Q and 224G of all Bangladeshi H5N1 isolates remained unchanged, suggesting an α-2,3-linked sialic acid preference and a greater likelihood of avian infectivity.Citation28 In the HA receptor–binding site, isolate A/Ck/BD/15210/12 (H5N1) had a single change within the 220-loop (217–224) K218R.
In the HA of the clade 2.2.2 isolates from Bangladesh, there were six glycosylation sites at positions 10 (NNST), 23 (NVT), 165 (NNT), 286 (NSS), 484 (NGS) and 543 (NGS) (H5 numbering). In addition to these glycosylation sites, clade 2.3.2.1 isolates also had another glycosylation site at position 140 (NSS). Isolate A/Ck/BD/18248/12 (H5N1) had loss of a glycosylation site at position 165, which was caused by the amino-acid substitution T167I.
NA
All the Bangladeshi H5N1 viruses had a 20-amino-acid deletion (positions 49–68) in the NA stalk region, which is reported to be required for influenza viruses to adapt from wild aquatic birds to domestic chickensCitation29 and for enhanced virulence in mice.Citation30 Amino acids 116V, 119E, 136Q, 156R, 199D, 223I, 247S, 275H, 277E and 295N (N2 numbering) in the NA conferred sensitivity to oseltamivir and/or zanamivir in the Bangladeshi H5N1 isolates.Citation31,Citation32,Citation33,Citation34 Isolate A/Dk/BD/18949/12 (H5N1) had the amino-acid substitution I117V, which may cause reduced susceptibility to oseltamivir.Citation32 Most of the isolates had NA 149V, suggesting sensitivity to zanamivir,Citation35 but we also found two mutations 149I (A/Ck/BD/18248/12 (H5N1)) and 149F (A/Env/BD/19338/13 (H5N1)) at that position that had not been previously reported to cause resistance.
PB2
The PB2 E627K mutation, which is important for mammalian host adaptation in AIVs,Citation36 was present in the H5N1 isolates from clade 2.2.2, but not in any of the viruses from clade 2.3.2.1 isolated in Bangladesh, India and Nepal. The presence of amino-acid substitutions L89V, G309D, R477G, I495V and A676T of the PB2 protein in all H5N1 viruses isolated in this study suggests enhanced polymerase activity and increased virulence in mice.Citation37
The PB2 amino-acid residue 702R, which was present in most of the H5N1 viruses from Bangladesh and India isolated in 2011 or later (clade 2.3.2.1) is a molecular marker of human H1N1, H2N2 and H3N2 and classical swine isolates and was previously found in the 1918 pandemic virus, A/PR/8/34 (H1N1) and seasonal human isolates.Citation38 Isolates of avian and equine origin usually possess PB2 amino-acid residue 702K.
PB1
The isolate A/Dk/BD/18949/12 (H5N1) had the PB1 amino-acid substitution I368V. This mutation may be important for transmission of H5 viruses in ferrets,Citation1 and has been observed in the PB1 protein sequence of most of the newly emerged H7N9 viruses in eastern China and H9N2 viruses from China.Citation39
PA
Another human-like mutation found in clade 2.3.2.1 H5N1 avian isolates from Bangladesh and India was PA S421I, which has been previously reported in A/Vietnam/1203/2004 (H5N1). In A/Ck/Vietnam/NCVD5/2003 (H5N1), this mutation in combination with PB2 E627K causes increased pathogenicity, higher replication titers and higher mortality in mice.Citation40
M
The M1 proteins of isolates A/Ck/BD/15079/12 (H5N1) and A/Ck/BD/15083/12 (H5N1) possess several hallmark amino-acid residues of the recently emerged human and avian H7N9 viruses and of H9N2 viruses from China (L46I, T140A, V142G, I219V, A227T, K242N and M248L). None of these residues was present in the Bangladeshi H9N2 isolates. In the M2 protein, isolate A/Ck/BD/15078/12 (H5N1) had the V27A mutation and isolates A/Ck/BD/15079/12 (H5N1) and A/Ck/BD/15083/12 (H5N1) had the S31N mutation. Both mutations are known to cause reduced susceptibility to amantadine and rimantadine.Citation41
NS
All Bangladeshi H5N1 isolates from clades 2.2.2 and 2.3.2.1 had a 5-amino-acid deletion (positions 80–85) in the NS protein, which is also present in other HPAI H5N1 viruses isolated after 2001 and is associated with increased virulence of H5N1 viruses in chickens and mice.Citation42
NP
Amino-acid changes in the NP genes of the Bangladeshi H5N1 viruses from clade 2.3.2.1 found also in isolates from India are shown in .
Phylogenetic analysis of Bangladeshi H5N1 isolates
To track the evolutionary relationships of the H5N1 viruses isolated from Bangladesh with viruses representing different H5N1 clades and lineages, as well as other influenza viral subtypes, clade affiliations of all viruses included in the study were assigned in the HA phylogenetic tree and then used in the phylogenetic trees of the other seven genes.
HPAI H5N1 viruses from clade 2.2.2
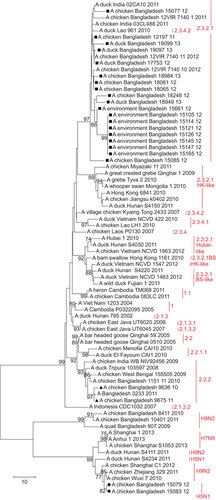
Phylogenetic analysis of the HA gene of 30 H5N1 isolates from Bangladesh showed that two isolates—A/Ck/BD/9636/10 (H5N1) and A/Ck/BD/9675/11 (H5N1)—belonged to clade 2.2.2 (Figure ). In the phylogenetic trees of all eight genes both isolates clustered together and were closely related to H5N1 viruses from clade 2.2.2 isolated in Bangladesh in 2010 and 2011, which all descended from H5N1 isolates from West Bengal and Tripura in India from 2008 and 2009. Their common ancestors were H5N1 isolates from bar-headed geese from Qinghai Lake in 2005 (– and Supplementary Figures S1–S4). The closest relative of isolate A/Ck/BD/9675/11 (H5N1) in all genes was isolate A/Bangladesh/3233/2011 (H5N1), which showed 99% similarity in the basic local alignment search tool (BLAST) algorithm analysisCitation43 in NCBI.
Figure 2 Phylogenetic tree of the HA genes of HPAI H5N1 viruses from Bangladesh, generated by neighbor-joining method in MEGA 5. Numbers at the branches indicate bootstrap values; only values >70 are shown. ▴ indicates Bangladesh isolates clade 2.2.2; ▪ indicates Bangladesh isolates from the A/Hubei/1/10-like lineage of clade 2.3.2.1; • indicates Bangladesh isolates from the A/Hong Kong/6841/10-like lineage of clade 2.3.2.1.
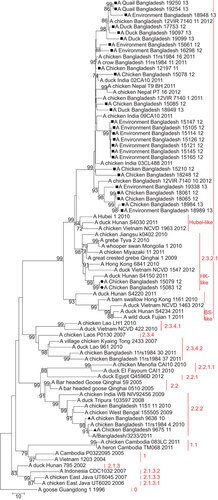
Figure 3 Phylogenetic tree of the NA gene of H5N1 viruses from Bangladesh, generated by neighbor-joining method in MEGA 5. Numbers at the branches indicate bootstrap values; only values >70 are shown. ▴ indicates Bangladesh isolates clade 2.2.2; ▪ indicates Bangladesh isolates from the A/Hubei/1/10-like lineage of clade 2.3.2.1; • indicates Bangladesh isolates from the A/Hong Kong/6841/10-like lineage of clade 2.3.2.1.
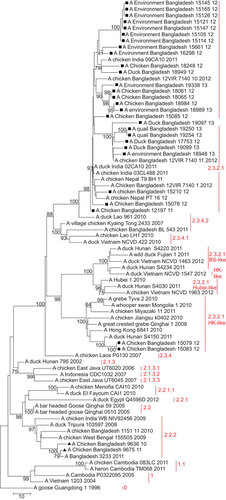
Figure 4 Phylogenetic tree of the PB2 gene of H5N1 viruses from Bangladesh, generated by neighbor-joining method in MEGA 5. Numbers at the branches indicate bootstrap values; only values >70 are shown. ▴ indicates Bangladesh isolates clade 2.2.2; ▪ indicates Bangladesh isolates from the A/Hubei/1/10-like lineage of clade 2.3.2.1; • indicates Bangladesh isolates from the A/Hong Kong/6841/10-like lineage of clade 2.3.2.1.
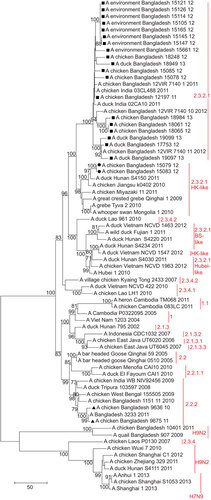
Figure 5 Phylogenetic tree of the M gene of H5N1 viruses from Bangladesh, generated by neighbor-joining method in MEGA 5. Numbers at the branches indicate bootstrap values; only values >70 are shown. ▴ indicates Bangladesh isolates clade 2.2.2; ▪ indicates Bangladesh isolates from the A/Hubei/1/10-like lineage of clade 2.3.2.1; • indicates Bangladesh isolates from the A/Hong Kong/6841/10-like lineage of clade 2.3.2.1.
HPAI H5N1 viruses from clade 2.3.2.1
Of the 30 H5N1 isolates in this study, 28 (93%) fell within clade 2.3.2.1 of the HA phylogenetic tree and 26 (86.7%) clustered together in one distinct group of the phylogenetic tree, which also included viruses from India identified in 2011 and from Nepal identified in 2011 and 2012. This monophyletic cluster did not change as a group in the phylogenetic analysis of the eight gene segments and showed evolutionary distance from the closest relatives outside the group. The cluster shared a common node with the A/Hubei/1/10-like genetic group in the HA, PB1 and NS trees (Figure and Supplementary Figures S1 and S4). In the PA gene tree (Supplementary Figure S2), the Bangladeshi clade 2.3.2.1 shared a node with a group of clade 2.3.2.1 viruses, which included all representatives of the A/Hubei/1/10-like lineage as well viruses of the A/HK/6841/10-like and the A/Barn swallow/HK/1161/10-like lineages. In the PB2 tree (Figure ), the big cluster of Bangladeshi clade 2.3.2.1 viruses shared a common node with the A/HK/6841/10-like lineage of clade 2.3.2.1. Interestingly, in the NA (Figure ) and NP gene (Supplementary Figure S3) trees, the Bangladeshi clade 2.3.2.1 cluster was a descendent of A/Dk/Lao/961/10 (H5N1), clade 2.3.4.2, and was clearly distinct from all 3 lineages of clade 2.3.2.1 (A/Hubei/1/10-like, A/HK/6841/10-like and A/BS/HK/1161/10-like lineages).
In the M gene tree (Figure ), the Bangladeshi cluster of clade 2.3.2.1 viruses also included A/Dk/Lao/961/10 (H5N1), clade 2.3.4.2. In the phylogenetic tree built using the neighbor-joining method, the cluster shared a node with the clade 2.3.2.1 viruses from the A/HK/6841/10-like lineage (Figure ). However, in the tree built using the maximum likelihood method (not shown), the cluster shared a node with clade 2.3.4.2. In both cases, clade 2.3.4 was the ancestor of the Bangladeshi H5N1 2.3.2.1 viruses with regard to the M gene. Thus, the results of the phylogenetic analysis suggest that the majority of the currently circulating H5N1 viruses from clade 2.3.2.1 in Bangladesh have undergone intraclade reassortment in the PB2 gene with 2.3.2.1 H5N1 viruses genetically close to the A/HK/6841/10-like lineage, and interclade reassortment in the NA, NP and M genes with H5N1 viruses from clade 2.3.4.2.
Since this cluster of Bangladeshi clade 2.3.2.1 viruses is very homogenous (i.e., it always has the same virus pattern in all trees) and shows that all isolates included have the same ancestors, this may be a sign of a single introduction of clade 2.3.2.1 in 2011 into the geographic region of Bangladesh, India and Nepal and ongoing evolution within the region that is supported by poultry movement. In all the phylogenetic trees in this study, some taxa within the Bangladeshi 2.3.2.1 cluster were not strongly supported by the bootstrap value (<70%), which is indicative of an ongoing evolutionary process.
Within the HA phylogenetic tree, isolates A/Ck/BD/15079/12 (H5N1) and A/Ck/BD/15083/12 (H5N1), both original samples collected on the same day in January 2012 from the same live bird market in Dhaka city, fell within clade 2.3.2.1, but did not cluster together with the rest of the H5N1 isolates from Bangladesh. Both isolates had identical HA nucleotide sequences and were genetically related to A/duck/Hunan/S4150/11 (H5N1). They all fell within the A/Hong Kong/6841/2010 (H5N1)-like genetic lineage of clade 2.3.2.1. This genetic group mainly has H5N1 viruses from Japan, China, Vietnam and Indonesia (Figure ).
Detection of reassortant H5N1 viruses with an H9N2-like M gene
The A/HK/6841/10 (H5N1)-like viruses A/Ck/BD/15079/12 (H5N1) and A/Ck/BD/15083/12 (H5N1) were closely related to A/duck/Hunan/S4150/11 (H5N1) in the H5, N1, PB2, PA, NP and NS gene phylogenetic trees and closely related to A/duck/Hunan/S4150/11 (H5N1) and A/duck/Hunan/S4220/11 (H5N1) in the PB1 phylogenetic tree. However, phylogenetic analysis of the M gene revealed that the M genes of isolates A/Ck/BD/15079/12 (H5N1) and A/Ck/BD/15083/12 (H5N1) were derived from H9N2 viruses isolated in eastern China and are closely related to A/Chicken/Wuxi/7/10 (H9N2) and A/Chicken/Zhejiang/329/11 (H9N2) (Figure ). Nucleotide BLAST analysis in NCBI of the M gene sequence of isolates A/Ck/BD/15079/12 (H5N1) and A/Ck/BD/15083/12 (H5N1) showed maximum identity of 99% with the M gene of A/Chicken/Rizhao/1313/13 (H9N2) and some other H9N2 viruses isolated in 2013 from the same location, as well as A/Chicken/Hong Kong/JV75/11 (H9N2), A/Chicken/Zhejiang/329/11 (H9N2), and isolates from Shanghai and other locations in eastern China.
Many H9N2 viruses were isolated from the same live bird markets in Dhaka city during the last few years, but we did not detect reassortment between the H5N1 and H9N2 lineages circulating in Bangladesh. The Bangladeshi H9N2 isolates represented the G1 genetic lineage and in the M gene phylogenetic tree, they fell within an evolutionarily distinct cluster from the H9N2 isolates from China (Figure ). The M gene phylogenetic tree also revealed that the M gene of isolates A/Ck/BD/15079/12 (H5N1) and A/Ck/BD/15083/12 (H5N1) and of recently emerged H7N9 viruses isolated from humans and birds in China have common ancestors—M genes of AI H9N2 viruses circulating in China. Negovetich et al.Citation15 reported in 2011 that influenza viruses from subtypes H1, H3, H4 and H10 have been isolated from poultry markets in Bangladesh and we detected LPAI H3N8 in a duck farm in 2012; however, reassortment between any of the abovementioned LPAI subtypes and H5N1 was not detected.
Discussion
Bangladesh experienced its first HPAI H5N1 outbreak in February 2007, and the isolated viruses belonged to clade 2.2 of the Qinghai lineage that emerged in 2005.Citation44 Phylogenetic analysesCitation44 and evidence of wild-bird migrationCitation45 suggested that the virus had been introduced into Bangladesh by migratory birds from Mongolia in early 2007. Since then, the HPAI H5N1 virus has become endemic to the poultry in Bangladesh.
In January 2011, clade 2.3.2.1 was introduced into the country. Islam et al.Citation11 reported the first cases of the new clade in a crow, a duck and a quail, and Khan et al.Citation46 reported a crow die-off caused by clade 2.3.2.1 in Bangladesh, which had started on 17 January 2011.
Phylogenetic analyses of the earliest clade 2.3.2.1 isolates from Bangladesh revealed a close relationship with H5N1 viruses from Nepal from 2010 and India from February 2011, with wild birds from Mongolia and Tyva from 2009 to 2011 being the common ancestors for all these viruses.Citation11,Citation47 Although the exact pathway of introduction of clade 2.3.2.1 into Bangladesh remains unclear, it is possible that virus transmission to neighboring territories occurred through crows, after the virus had been already introduced into Nepal from Mongolia and Tyva via migratory birds previously. Clade 2.3.2.1 is currently circulating in poultry in Bangladesh and has displaced clade 2.2.2.
H5N1 viruses from clade 2.3.4.2 were isolated in Bangladesh in February and March 2011,Citation10,Citation11,Citation47 but these viruses were only sporadically detected and did not establish a sustained circulation.
We report here the isolation of 66 HPAI H5N1 viruses and further analyses of 2 H5N1 viruses from clade 2.2.2 from December 2010 and January 2011 and of 28 H5N1 isolates from clade 2.3.2.1 from 2011 to 2013. Until February 2011, we only detected H5N1 viruses from clade 2.2.2. During March–July 2011, we did not detect any H5N1 viruses in the samples from Bangladesh, and from August 2011 to 2013 the only H5N1 clade detected was 2.3.2.1, indicating that the clade change occurred during the latter period.
All HPAI H5N1 viruses and 99% of the H9N2 viruses isolated in this study were detected at the live bird markets in Dhaka city, which underlines the important role that the markets play in perpetuating and maintaining both subtypes. We did not detect H5N1 viruses on the farms, which may not be representative of all farms in the country. During the five years of surveillance, we did not observe any clinical signs of a disease at the farms and retail markets in Bangladesh. Normal bird mortality rates were reported by poultry workers and retailers at the farms and markets where we were collecting samples. The lack of morbidity and mortality among poultry is unusual for HPAI H5N1 viruses, and may be explained with protection against the HPAI H5N1 viruses provided by the year-round circulating LPAI H9N2 viruses. However, this hypothesis has not been tested because it was beyond the scope of our study.
In January 2012, we detected two reassortant (7:1) HPAI (H5N1) clade 2.3.2.1 viruses that were genetically distinct from all the other viruses isolated from Bangladesh and possessed the M gene from the H9N2 subtype, which circulated in China in 2010–2012. The remaining seven genes of isolates A/Ck/BD/15079/12 (H5N1) and A/Ck/BD/15083/12 (H5N1) were closely related to A/HK/6841/10-like A/duck/Hunan/S4150/11 (H5N1) from clade 2.3.2.1. The phylogenetic analysis revealed that not only the reassortant H5N1 viruses from Bangladesh but also A/duck/Hunan/S4111/11 (H9N2) and A/duck/Hunan/S4234/11 (H5N1) fall within the Chinese H9N2 cluster of the M gene tree. All the three above-mentioned duck viruses from Hunan were isolated in November 2011 from a backyard-style duck farm with an open environment and no biosecurity measures that was located in the Dongting Lake region of China.Citation48 This lake is the third biggest lake in China and is on the East Asian–Australasian Flyway of migratory birds. Therefore, on the basis of the finding that none of the genes from reassortant H5N1 viruses originated from local isolates, we hypothesize that these viruses had been directly introduced into Bangladesh either via migratory birds, using overlapping flyways from the Dongting region in China to Bangladesh, or via poultry trade and movement, combined with infected wild-bird migration. This reassortant 7:1 H5N1 genotype was detected only once in Bangladesh, representing transient infection in chickens. Our surveillance results provide further confirmation that the H9N2 viruses circulating in China have been involved in multiple reassortment events with both LPAI and HPAI viruses, but with different outcomes. Although the reassortant viruses reported in this study did not affect the H5N1 situation in Bangladesh, the newly emerged H7N9 viruses in China caused many human deaths in 2013.
Another important finding of this study is that despite many years of cocirculation of H5N1 and H9N2 viruses in Bangladesh, we did not find reassortant H5N1 viruses that possessed internal genes from the local H9N2 lineage. This finding is surprising, because in this study H5N1 viruses were more often detected as a co-infection with H9N2 than alone (66 H5N1 isolates versus 78 isolates containing both H5N1 and H9N2 in a single-host sample). Shanmuganatham et al.Citation16 also found no H9N2 viruses from Bangladesh that possessed internal genes from H5N1. As of November 2013, one study has reported two reassortant H5N1 viruses with the H9N2 PB1 gene in poultry in Bangladesh.Citation49 Thus, reassortant viruses between the H5N1 and H9N2 subtypes apparently occur in Bangladesh, but our surveillance suggests that they do not have the fitness to supplant the dominant clade 2.3.2.1. This may be because the H9N2 lineage that currently circulates in Bangladesh is a reassortant virus possessing three gene segments from subtype H7N3.Citation16
Our molecular and phylogenetic analyses of the H5N1 viruses showed that the majority of H5N1 isolates from Bangladesh from both clade 2.2.2 and clade 2.3.2.1 were genetically similar to and clustered together with contemporary H5N1 isolates from India and Nepal. The viruses from clade 2.2.2 did not show any evidence of genetic reassortment. They seemed to have established a stable lineage for the period of their circulation in Bangladesh (February 2007–February 2011) without reassorting with other subtypes.
Of the H5N1 viruses from clade 2.3.2.1, 93% had common ancestry and formed an evolutionary distinct ‘Bangladesh’ cluster in all eight gene phylogenetic trees. By conducting full genome phylogenetic analyses, we could detect intra- and inter-clade reassortment events that occurred in the Bangladeshi clade 2.3.2.1 isolates and those from India and Nepal. The HA, PB1, NS and PA genes had common ancestors with the A/Hubei/1/10-like (H5N1) 2.3.2.1 lineage, whereas the PB2 genes were similar to A/HK/6841/10-like (H5N1) 2.3.2.1 viruses. Even more interesting was the finding that the NA, NP and M genes were clearly related to clade 2.3.4.2. This clade was detected in the Chittagong district in East Bangladesh in February–March 2011 when clade 2.3.2.1 had already been introduced into the country, but it circulated for a short period of time and did not spread to other districts. Because there is limited sequence information for clade 2.3.4.2 from Bangladesh in public databases, we could not conclude whether clade 2.3.4.2 from Bangladesh had been involved in the reassortment events of clade 2.3.2.1, especially with regard to internal genes. In the NA tree, the only clade 2.3.4.2 virus from Bangladesh (A/Ck/Bangladesh/BL/543/11 (H5N1)) seems to be genetically related to viruses from Kyang Tong (Myanmar), Vietnam and Laos, but in a cluster that is distinct from A/Dk/Lao/961/10, which shares a node with the Bangladeshi clade 2.3.2.1 isolates. Thus, it is possible that the H5N1 clade 2.3.2.1 had been introduced in the region of Bangladesh, India and Nepal after the reassortment events involving clade 2.3.4.2 occurred. This theory would explain why the majority of isolates from the region have the same gene constellation.
Our overall results from the phylogenetic analyses suggest that both clades 2.2.2 and 2.3.2.1 might have had one initial introduction each into the region by migratory birds, followed by an intensive evolutionary process driven by poultry movements and cross-border viral transmission among Bangladesh, India and Nepal. New H5N1 viruses were introduced into Bangladesh either via wild-bird migrations or by poultry trade, but they have not persisted in the poultry population. The molecular analysis of the H5N1 viruses isolated in Bangladesh showed that they share many common molecular changes in all eight genes with isolates from India and Nepal, forming a clade-specific genetic signature of the H5N1 viruses that evolved within this region. This is valid for both clades 2.2.2 and 2.3.2.1.
Also, some human-like mutations were found in the genome of the H5N1 viruses circulating in Bangladesh, which might explain the low number of human H5N1 cases reported in the country. In contrast to clade 2.2.2, the currently circulating clade 2.3.2.1 does not carry the PB2 E627K mutation, which is important for mammalian host adaptation. The pathogenicity role of the two human-like mutations PB2 K702R and PA S421I, which are a part of the genetic signature of the clade 2.3.2.1 H5N1 viruses from Bangladesh and surrounding regions and have previously been described in the 1918 pandemic virus and in the A/Vietnam/1203/2004 (H5N1), respectively, needs to be further investigated.
Several mutations found in the Bangladeshi isolates have been previously reported to cause binding to α-2,6 receptors and increased pathogenicity in mice.Citation23,Citation24,Citation25 Molecular analysis showed that the NA protein of one of the recent isolates had the I117V mutation, which may cause reduced susceptibility to the commonly used neuraminidase inhibitor oseltamivir. Also, three other viruses from 2012, including the reassortant H5N1 isolates, had M2 protein mutations V27A and S31N, which can cause reduced susceptibility to the M2 ion channel blockers amantadine and rimantadine.
Our HI assays showed that all tested H5N1 isolates from the currently circulating Bangladeshi clade 2.3.2.1 isolated until 2013 are antigenically homogenous. Further surveillance activities are required to clarify whether the lower titer of the most recent clade 2.3.2.1 isolates is due to antigenic drift. The variety of host bird species (chickens, ducks and quail) of the H5N1 viruses in Bangladesh did not lead to antigenic diversity. Although the HA glycoprotein of the majority of the currently circulating clade 2.3.2.1 viruses is genetically closely related to that of the Hubei/1/10-like lineage of clade 2.3.2.1, antigenically, they do not show cross-reactivity with this virus. Because of the poor reactivity of the Bangladeshi Hubei/1/10-like clade 2.3.2.1 viruses to the prototype virus, the World Health Organization recently chose the isolate A/duck/Bangladesh/19097/2013 (H5N1) reported in this study as a new candidate vaccine virus to represent the H5N1 lineage circulating in this region.Citation4
The results of our study emphasize the need of continuous surveillance activities in Bangladesh that would allow detection of any newly emerged AIV with pandemic potential.
Supplementary information
Download PDF (2.6 MB)Supplementary information
Download PDF (131.6 KB)Supplementary information
Download PDF (130.6 KB)Supplementary information
Download PDF (2.6 MB)We thank Jerry Parker and Richard Elia for maintaining the influenza database at St Jude Children’s Research Hospital, James Knowles (St Jude Children's Research Hospital) for providing administrative assistance, Klo Spelshouse (St Jude Children's Research Hospital) for assistance with the figures and Vani Shanker (St Jude Children's Research Hospital) for editing this manuscript. This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (contract HHSN272200900007C); and the American Lebanese Syrian Associated Charities.
Supplementary information
Note: Supplementary information for this article can be found on Emerging Microbes & Infections' website (http://www.nature.com/emi/).
- Herfst S, Schrauwen EJ, Linster M et al.Airborne transmission of influenza A/H5N1 virus between ferrets. Science2012;336: 1534–1541.
- Imai M, Watanabe T, Hatta M et al.Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature2012;486: 420–428.
- WHO/OIE/FAO H5N1 Evolution Working Group.Continued evolution of highly pathogenic avian influenza A (H5N1): updated nomenclature. Influenza Other Respir Viruses2012;6: 1–5.
- World Health Organization. Antigenic and genetic characteristics of A(H5N1), A(H7N3), A(H9N2) and variant influenza viruses and candidate vaccine viruses developed for potential use in human vaccines (n.d.).Geneva: WHO, 2013.Available at https://doi.org/http://www.who.int/influenza/vaccines/virus/characteristics_virus_vaccines/en/ (accessed 1 November 2013).
- Sakoda Y, Sugar S, Batchluun D et al.Characterization of H5N1 highly pathogenic avian influenza virus strain isolated from migratory waterfowl in Mongolia on the way back from the southern Asia to their northern territory. Virology2010;406: 88–94.
- Reid SM, Shell WM, Barboi G et al.First reported incursion of highly pathogenic notifiable avian influenza A H5N1 viruses from clade 2.3.2 into European poultry. Transbound Emerg Dis2011;58: 76–78.
- Marinova-Petkova A, Georgiev G, Seiler P et al.Spread of influenza virus A (H5N1) clade 2.3.2.1 to Bulgaria in common buzzards. Emerg Infect Dis2012;18: 1596–1602.
- The World Bank. Implementation completion and results report (IDA-43400 TF-90662) on a credit in the amount of SDR 10.5 million (US$16.0 million equivalent) to the People’s Republic of Bangladesh for an avian influenza preparedness and response project under the global program for avian influenza and human pandemic preparedness and response.Washington, DC: The World Bank, 2013.Available at: https://doi.org/http://www-wds.worldbank.org/external/default/WDSContentServer/WDSP/IB/2013/07/04/000442464_20130704100805/Rendered/PDF/ICR21770ICR0Av0Box0377341B00PUBLIC0.pdf (accessed 17 October 2013).
- World Organization for Animal Health.Update on highly pathogenic avian influenza in animals (type H5 and H7), 2013, Bangladesh follow-up report No. 42.Paris: OIE, 2013.Available at: https://doi.org/http://www.oie.int/animal-health-in-the-world/update-on-avian-influenza/2013 (accessed on 13 November 2013).
- Food and Agriculture Organization Emergency Prevention System. H5N1 global overview, July–September 2011.Rome: FAO, 2011.Available at: https://doi.org/http://www.fao.org/docrep/015/al913e/al913e00.pdf (Accessed 13 November 2013).
- Islam MR, Haque ME, Giasuddin M et al.New introduction of clade 2.3.2.1 avian influenza virus (H5N1) into Bangladesh. Transbound Emerg Dis2012;59: 460–463.
- World Health Organization. Situation updates—avian influenza.Geneva: WHO, 2013.Available at https://doi.org/http://www.who.int/influenza/human_animal_interface/avian_influenza/archive/en/index.html (accessed 13 November 2013).
- Brooks WA, Alamgir A, Sultana R et al.Avian influenza virus A (H5N1) detected through routine surveillance in child, Bangladesh. Emerg Infect Dis2009;15: 1311–1313.
- International Centre for Diarrhoeal Diseases Research, Bangladesh. The first fatal human infection with highly pathogenic avian influenza A (H5N1) virus detected in Bangladesh. Health and Science Bulletin2013;11: 1–6.
- Negovetich NJ, Feeroz MM, Jones-Engel L et al.Live bird markets of Bangladesh: H9N2 viruses and the near absence of highly pathogenic H5N1 influenza. PLoS ONE2011;6: e19311.
- Shanmuganatham K, Feeroz MM, Jones-Engel L et al.Antigenic and molecular characterization of avian influenza A(H9N2) viruses, Bangladesh. Emerg Infect Dis2013;19: 1393–1402.doi: https://doi.org/10.3201/eid1909.130336.
- World Health Organization. CDC protocol of real time RTPCR for swine influenza A (H1N1), 28 April 2009.Geneva: WHO, 2009.Available at https://doi.org/http://www.who.int/csr/resources/publications/swineflu/CDCrealtimeRTPCRprotocol_20090428.pdf (accessed 13 November 2013).
- Palmer DFDW, Coleman MT, Schild GC. Advanced laboratory techniques for influenza diagnosis. Washington, DC: US Department of Health, Education and Welfare, 1975.
- Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR.Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol2001;146: 2275–2289.
- National Center for Biotechnology Information. Influenza virus sequence database.Bethesda: NCBI, 2013.Available at https://doi.org/http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi?go=database (accessed 13 November 2013).
- Global Initiative on Sharing All Influenza Data. EpiFlu Database.Munich: GISAID, 2013.Available at https://doi.org/http://platform.gisaid.org (accessed13 November 2013).
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S.MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol2011;28: 2731–2739.
- Yen HL, Aldridge JR, Boon AC et al.Changes in H5N1 influenza virus hemagglutinin receptor binding domain affect systemic spread. Proc Natl Acad Sci USA2009;106: 286–291.
- Stevens J, Blixt O, Tumpey TM et al.Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science2006;312: 404–410.
- Su Y, Yang HY, Zhang BJ, Jia HL, Tien P.Analysis of point mutation in H5N1 avian influenza virus hemagglutinin in relation to virus entry into live mammalian cells. Arch Virol2008;153: 2253–2261.
- Horimoto T, Kawaoka Y.Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza A virus. J Virol1994;68: 3120–3128.
- Chen H, Li Y, Li Z et al.Properties and dissemination of H5N1 viruses isolated during an influenza outbreak in migratory waterfowl in western China. J Virol2006;80: 5976–5983.
- Ha Y, Stevens DJ, Skehel JJ, Wiley DC.X-ray structures of H5 avian and H9 swine influenza virus hemagglutinins bound to avian and human receptor analogs. Proc Natl Acad Sci USA2001;98: 11181–11186.
- Matrosovich M, Zhou N, Kawaoka Y, Webster RG.The surface glycoproteins of H5 influenza viruses isolated from humans, chickens, and wild aquatic birds have distinguishable properties. J Virol1999;73: 1146–1155.
- Zhou H, Yu Z, Hu Y et al.The special neuraminidase stalk-motif responsible for increased virulence and pathogenesis of H5N1 influenza A virus. PLoS ONE2009;4: e6277.
- Hurt AC, Holien JK, Barr IG.In vitro generation of neuraminidase inhibitor resistance in A(H5N1) influenza viruses. Antimicrob Agents Chemother2009;53: 4433–4440.
- Ilyushina NA, Seiler JP, Rehg JE, Webster RG, Govorkova EA.Effect of neuraminidase inhibitor-resistant mutations on pathogenicity of clade 2.2 A/Turkey/15/06 (H5N1) influenza virus in ferrets. PLoS Pathog2010;6: e1000933.
- Govorkova EA, Ilyushina NA, Boltz DA, Douglas A, Yilmaz N, Webster RG.Efficacy of oseltamivir therapy in ferrets inoculated with different clades of H5N1 influenza virus. Antimicrob Agents Chemother2007;51: 1414–1424.
- Ilyushina NA, Bovin NV, Webster RG.Decreased neuraminidase activity is important for the adaptation of H5N1 influenza virus to human airway epithelium. J Virol2012;86: 4724–4733.
- Naughtin M, Dyason JC, Mardy S et al.Neuraminidase inhibitor sensitivity and receptor-binding specificity of Cambodian clade 1 highly pathogenic H5N1 influenza virus. Antimicrob Agents Chemother2011;55: 2004–2010.
- Le QM, Sakai-Tagawa Y, Ozawa M, Ito M, Kawaoka Y.Selection of H5N1 influenza virus PB2 during replication in humans. J Virol2009;83: 5278–5281.
- Li J, Ishaq M, Prudence M et al.Single mutation at the amino acid position 627 of PB2 that leads to increased virulence of an H5N1 avian influenza virus during adaptation in mice can be compensated by multiple mutations at other sites of PB2. Virus Res2009;144: 123–129.
- Taubenberger J, Reid A, Lourens R, Wang R, Jin G, Fanning T.Characterization of the 1918 influenza virus polymerase genes. Nature2005;437: 889–893.
- Wang Y, Dai Z, Cheng H et al.Towards a better understanding of the novel avian-origin H7N9influenza A virus in China. Sci Rep2013;3: 2318.
- Kim JH, Hatta M, Watanabe S, Neumann G, Watanabe T, Kawaoka Y.Role of host-specific amino acids in the pathogenicity of avian H5N1 influenza viruses in mice. J Gen Virol2010;91: 1284–1289.
- Ilyushina NA, Govorkova EA, Webster RG.Detection of amantadine-resistant variants among avian influenza viruses isolated in North America and Asia. Virology2005;341: 102–106.
- Long JX, Peng DX, Liu YL, Wu YT, Liu XF.Virulence of H5N1 avian influenza virus enhanced by a 15-nucleotide deletion in the viral nonstructural gene. Virus Genes2008;36: 471–478.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.Basic local alignment search tool. J Mol Biol1990;215: 403–410.
- Ahmed SS, Themudo GE, Christensen JP, Biswas PK, Giasuddin M, Samad MA.Molecular epidemiology of circulating highly pathogenic avian influenza (H5N1) virus in chickens, in Bangladesh, 2007–2010. Vaccine2012;30: 7381–7390.
- Gilbert M, Newman SH, Takekawa JY et al.Flying over an infected landscape: distribution of highly pathogenic avian influenza H5N1 risk in South Asia and satellite tracking of wild waterfowl. Ecohealth2010;7: 448–458.
- Khan SU, Berman L, Haider N et al.Investigating a crow die-off in January–February 2011 during the introduction of a new clade of highly pathogenic avian influenza virus H5N1 into Bangladesh. Arch Virol.1 October 2013.doi: https://doi.org/10.1007/s00705-013-1842-0.
- Mondal S, Balasuriya U, Yamage M.Genetic diversity and phylogenetic analysis of highly pathogenic avian influenza (HPAI) H5N1 viruses circulating in Bangladesh from 2007–2011. Transbound Emerg Dis.11 October 2013.doi: https://doi.org/10.1111/tbed.12173.
- Dong G, Tan D, Shi J et al.Complex reassortment of multiple subtypes of avian influenza viruses in domestic ducks at the Dongting Lake region in China. J Virol2013;87: 9452–9462.
- Monne I, Yamage M, Dauphin G et al.Reassortant avian influenza A(H5N1) viruses with H9N2-PB1 gene in poultry, Bangladesh. Emerg Infect Dis2013;19: 1630–1634.