
Abstract
Leptospira santarosai serovar Shermani is the most frequently encountered serovar, and it causes leptospirosis and tubulointerstitial nephritis in Taiwan. This study aims to complete the genome sequence of L. santarosai serovar Shermani and analyze the transcriptional responses of L. santarosai serovar Shermani to renal tubular cells. To assemble this highly repetitive genome, we combined reads that were generated from four next-generation sequencing platforms by using hybrid assembly approaches to finish two-chromosome contiguous sequences without gaps by validating the data with optical restriction maps and Sanger sequencing. Whole-genome comparison studies revealed a 28-kb region containing genes that encode transposases and hypothetical proteins in L. santarosai serovar Shermani, but this region is absent in other pathogenic Leptospira spp. We found that lipoprotein gene expression in both L. santarosai serovar Shermani and L. interrogans serovar Copenhageni were upregulated upon interaction with renal tubular cells, and LSS19962, a L. santarosai serovar Shermani-specific gene within a 28-kb region that encodes hypothetical proteins, was upregulated in L. santarosai serovar Shermani-infected renal tubular cells. Lipoprotein expression during leptospiral infection might facilitate the interactions of leptospires within kidneys. The availability of the whole-genome sequence of L. santarosai serovar Shermani would make it the first completed sequence of this species, and its comparison with that of other Leptospira spp. may provide invaluable information for further studies in leptospiral pathogenesis.
Introduction
Leptospirosis is a re-emerging infectious zoonotic disease that occurs in tropical and subtropical regions.Citation1,Citation2,Citation3 In Taiwan, a major outbreak occurred in 2009 after typhoon Morakot, with 203 confirmed cases of leptospirosis.Citation4 Leptospirosis, as caused by leptospires, is characterized by fever, jaundice, renal failure and/or pulmonary hemorrhage culminating in multiple-organ dysfunction. Pathogenic Leptospira species are transmitted to humans after contact with animal reservoirs or through environmental contamination with their urine.Citation5,Citation6 Kidney injury is an early manifestation of acute leptospirosis, occurring within days of infection, and kidney damage occurs late in chronic infections.Citation7 Tubulointerstitial nephritis presents either in an acute or chronic form, and it is the primary cause of renal injury in leptospirosis. Leptospirosis is a common cause of acute tubulointerstitial nephritis that may cause acute kidney injury, and it has the propensity to damage blood vessels and the kidney structure.Citation8 During leptospiral chronic infection, tubulointerstitial nephritis is the most common lesion that can progress to fibrosis and subsequent renal failure. In kidneys, the leptospires chronically infect and harbor the bacteria in renal tubules.Citation7 The mechanisms of Leptospira kidney pathogenesis remain unclear, and the virulent factors of Leptospira need further identification.
Leptospira species belonging to multi-chromosomal genomes consist of a genetically diverse group of pathogenic, intermediate pathogenic and saprophytic species.Citation9,Citation10 L. interrogans contains a large number of serogroups, the strains of which are pathogenic for humans and animals, whereas L. bijlexa also contains a large number of serogroups, which are saprophytic species primarily found in fresh surface water and moist soil. Whole-genome sequences of Leptospira species are being completed, allowing for a comparative genomic analysis of the adaptation of different species to their natural habitats and pathogenesis.Citation11 To date, the whole-genome analysis of Leptospira species has provided insights into their pathogenesis, and genome sequencing efforts have so far focused on pathogenic (L. interrogans and L. borgpetersenii) and saprophytic species (L. biflexa).Citation12,Citation13,Citation14,Citation15,Citation16,Citation17,Citation18 The genome sequence of L. interrogans serovar Lai was published, and a comparative genomic analysis with L. interrogans serovar Copenhageni has been performed.Citation19 Our team has sequenced a draft genome of L. santarosai serovar Shermani, the highest prevalent serovar in Taiwan, by using high-throughput Illumina sequencing platforms.Citation20 The genome sequence of L. santarosai serovar Shermani has been deposited at DDBJ/EMBL/GenBank under the accession number ADOR00000000. However, a comparative genetic analysis based on BLASTx data revealed that only 73% of all coding sequences (CDS) include matches with pathogenic L. interrogans. These results suggested that L. interrogans and L. santarosai serovar Shermani might have different pathogenesis mechanisms. A recent report by Wilson et al.Citation21 indicated that the application of genome sequences may aid in the clinical diagnosis of neuroleptospirosis by L. santarosai; hence, the effort required to obtain the complete genome sequence is justified.
Several virulent factors in pathogenic Leptospira species have been identified, including lipopolysaccharide, lipoproteins, outer membrane proteins (OMPs) and cell wall components, which are functionally and structurally important for nutritional uptake, signal transduction, cell stabilization, and immunogenicity.Citation22,Citation23 Our previous finding indicates that a major OMP, namely LipL32 from L. santarosai serovar Shermani, can induce the secretion of inflammatory cytokines in murine renal tubular cells via a Toll-like receptor-dependent pathway and cause tubulointerstitial nephritis in mice.Citation24,Citation25,Citation26,Citation27 The major antigens of pathogenic Leptospira are lipopolysaccharide and lipoproteins, which are pathogen-associated molecular patterns found in the kidneys of Leptospira-infected animals, and they link to Leptospira-induced tubular interstitial nephritis.Citation28 Pathogenic leptospires have been shown to express OMPs and many lipoproteins, suggesting that they could play a role in kidney–pathogen interactions.
In this study, we described a method to assemble the complete genome sequence of L. santarosai serovar Shermani strain LT821 de novo by integrating second- and third- generation sequencing methods with optical whole-genome mapping. Here, we report the genome sequence of L. santarosai serovar Shermani strain LT821, a less-characterized bacterium in the genus Leptospira that has been linked to leptospirosis. The draft genome sequence consists of 111 contigs with a total determined size of 3 936 333 base pairs (bp) and 4033 predicted genes, and the majority of these non-orthologous genes encode hypothetical proteins.Citation20
To understand what characteristics differentiate these Leptospira species, particularly in terms of virulence capacity, we report the first genome sequence of L. santarosai and then perform a comparative genomic analysis between the whole-genome sequence and the recently described genome sequence of Leptospira species. In addition, L. santarosai serovar Shermani strain CCF, which was isolated from a Taiwanese patient with leptospirosis in 2001,Citation29 was analyzed for its gene sequences. Furthermore, comparative analyses of differential leptospiral gene expressions in L. santarosai serovar Shermani and L. interrogans serovar Copenhageni that infected human kidney 2 (HK-2) cells, which are human renal proximal tubular cells, were performed in this study. An analysis of leptospiral gene expression in cell-based infection models are vital for identifying differentially regulated genes that are relevant to pathogenesis.
The new features of the L. santarosai serovar Shermani found in this study may contribute to our understanding of the molecular mechanisms of leptospiral physiology and pathogenesis. The comparative genomic characteristics of this subset of human pathogens may contribute to our understanding of how they adapt to environments and acquire increased virulence.
MATERIALS AND METHODS
Bacterial strains, cell and culture conditions
L. santarosai serovar Shermani strain LT821 (ATCC number 43286) and L. interrogans serovar Copenhageni Fiocruz L1-130 (ATCC number BAA-1198) were purchased from the American Type Culture Collection (Manassas, VA, USA). A clinical L. santarosai serovar Shermani strain CCF was isolated from a Taiwanese patient with leptospirosis in 2010.Citation29 The bacteria were propagated at 28 °C under aerobic conditions in medium containing 10% Leptospira enrichment Ellinghausen–McCullough–Johnson–Harris medium (BD Diagnostics, Sparks, MD, USA) and 90% Leptospira medium base Ellinghausen–McCullough–Johnson–Harris (Difco, Sparks, MD, USA). Bacterial densities were counted with a CASY-Model TT cell counter and analyzer (Roche Innovatis AG, Casy-Technologh, Reutlingen, Germany). Leptospira genomic DNA was extracted from seven-day-old cultures by using a procedure similar to the cetyltrimethylammonium bromide precipitation method.Citation30 HK-2, the immortalized human renal proximal tubular cell line, was obtained from ATCC (number CRL-2190; Maryland, USA) and cultured in DMEM/Ham's F12 (Life Technologies, Paisley, UK) supplemented with 10% fetal calf serum (Biological Industries Ltd, Cumbernauld, UK), glutamine (Life Technologies, Paisley, UK), HEPES buffer (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; Gibco BRL, Paisley, UK), hydrocortisone, insulin, transferrin and sodium selenite (Sigma Chemical Company Ltd, Poole, UK). Cells were grown at 37 °C under a humidified atmosphere with 5% CO2. All experiments are performed under serum-free conditions to avoid the influence of serum on cell function and the investigated events.
Genome sequencing, assembly and annotation
The genomic DNA of L. santarosai serovar Shermani strain LT821 was subjected to high-throughput sequencing by using the next-generation sequencer: Illumina (Solexa) Genome Analyzer II DNA sequencer (Illumina Inc., San Diego, CA, USA), a 454 GS FLX platform (Roche, Branford, USA) and a Pacific Biosciences RS sequencer (the PacBio; Pacific Biosciences, Menlo Park, USA). The de novo assembly was performed by using Velvet, a hierarchical genome-assembly process (HGAP; Pacific Biosciences, Menlo Park, California, USA), A Hybrid Assembler (AHA; Pacific Biosciences, Menlo Park, California, USA) and the de novo assembler from CLC bio, a QIAGEN Company.Citation31,Citation32,Citation33 The order and orientation of these contigs were confirmed by optical mapping systems (OpGen Technologies Inc., Madison, WI, USA).Citation34 In brief, a whole-genome AflII map was constructed from randomly sheared L. santarosai serovar Shermani genomic DNA molecules digested with AflII. The map acted as a scaffold for the high-resolution whole-genome map was aligned with sequence contigs with MapManager software (OpGen Technologies Inc., Madison, WI, USA). The gap closure and validation of assembly sequences were achieved by polymerase chain reaction (PCR) and Sanger sequencing of the amplicons. We designed a subset of primer pairs that were located at a minimum distance of 50 bp upstream and downstream from the gaps and neighboring contig/scaffold ends. The genomic DNA of L. santarosai serovar Shermani was used as a template in the PCR reaction with an AccuPrime Taq DNA polymerase High Fidelity Kit (Invitrogen, Carlsbad, CA, USA) according to the following program: 95 °C for 1 min, then 35 cycles of 95 °C for 30 s, 65 °C for 30 s, 72 °C for 3 min followed by a final extension at 72 °C for 10 min. The successfully amplified products that did not contain nonspecific amplification products were recovered and purified by using a Gel/PCR DNA Fragments Extraction Kit (Geneaid Biotech Ltd., Taipei, Taiwan) and further studied by the DNA Sequencing Core Laboratory (Chang Gung Memorial Hospital, Linkou, Taiwan) by using Sanger sequencing in both forward and reverse directions. Assembly sequences were concatenated with CLC Genomics Workbench 5.1 (CLC Bio, Aarhus, Denmark) with default parameters. The finished assembled sequences were annotated from the Prokaryotic Genomes Automatic Annotation Pipeline (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html). Graphical maps of circular genomes were generated from the Circular Genome Viewer (CGView) Server.Citation35,Citation36
Comparative analysis
A comparison of whole genomes from L. santarosai serovar Shermani and previously published pathogenic Leptospira spp. was performed by the MAUVE alignment system, the CGView Comparison Tool and BLASTp analysis.Citation37 Previously published Leptospira spp. sequences were downloaded from the NCBI and the accession numbers of the genome sequences are listed in . Functional domains of putative proteins were identified by searching against the Pfam database.Citation38
Table 1 Genome features of Leptospira spp.
Nucleotide sequence accession number
The complete genome sequence of L. santarosai serovar Shermani strain LT821 (ATCC number 43286) has been deposited at DDBJ/EMBL/GenBank under the accession number CP006694 for chromosome I and at CP006695 for chromosome II.
The PCR-based identification of specific genes in a clinical L. santarosai serovar Shermani isolate from a patient with Leptospirosis
A PCR using primers designed from a L. santarosai serovar Shermani DNA sequence was used to investigate the presence of these genes in a clinical L. santarosai serovar Shermani strain CCF isolate from a Taiwanese patient with leptospirosis.Citation29 The primer sequences are listed in Supplementary Table S1. Leptospira genomic DNA (500 ng) was used as starting material. Standard Taq polymerase (1.25 units), 200 μM of each deoxynucleotide triphosphate and 0.5 μM primers were used. The amplification was performed in a PTC-100 Programmable Thermal Controller (M. J. Research Inc., Waltham, Massachusetts, USA) under the following conditions: 95 °C for 1 min for one cycle, 35 cycles of 95 °C for 30 s, 65 °C for 30 s and 72 °C for 3 min, followed by a final extension at 72 °C for 10 min. The PCR products were separated on 1% agarose gels containing 0.05% ethidium bromide and visualized under ultraviolet light, sized and photographed (ChemiDoc XRS system; Bio-Rad, Hercules, CA, USA). The amplification products were recovered and purified with a Gel/PCR DNA Fragments Extraction Kit (Geneaid Biotech. Ltd) and the sequencing of the resulting PCR products was performed by the DNA Sequencing Core Laboratory (Chang Gung Memorial Hospital, Linkou, Taiwan).
Cell-based infection models for leptospiral gene expression analysis
The cultured HK-2 cells were grown to 80%–90% confluence in fresh media without antibiotics and serum, and they were cultured for an additional 12 h before infection. Leptospira cells were harvested by centrifugation at 4000g for 15 min. The HK-2 cells in each sample were incubated in suspension with either L. santarosai serovar Shermani strain LT821 or L. interrogans serovar Copenhageni Fiocruz L1-130, or without any bacteria, for 4 h at 37 °C in 5% CO2. The multiplicity of infection was 100 bacteria per cell. After incubation, the cells were washed with PBS and harvested for RNA isolation. Each sample was lysed in 1 mL of RNA-Bee RNAzol reagent (Tel-Test Inc., Friendswood, TX, USA) with a DNaseI digestion according to the manufacturer's instructions. The total RNA concentrations were determined with a NanoDrop ND-1000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA). DNase-treated RNA (∼1 μg per sample) was reverse-transcribed with a First Strand cDNA Synthesis Kit for reverse transcription PCR (RT-PCR) (AMV) (Roche Diagnostics, Mannheim, Germany) with random primer p(dN)6s, dNTPs, 10x reaction buffer, MgCl2, RNase inhibitor and AMV reverse transcriptase. Quantitative real-time RT-PCR assays with SYBR Green PCR Master Mix (PE-Applied Biosystems, Cheshire, UK) were performed by using ABI ViiA7 real-time PCR systems (Applied Biosystems, Foster, CA, USA) for 50 cycles (95 °C 10 s, 60 °C 1 min and 60 °C 1 min). A dissociation curve step was added to ensure the optimization of the primers. For each primer pair described in Supplementary Table S1, no-template control reactions were employed and each reaction was performed in triplicate. The threshold cycle number (CT) was determined by using PE-Applied Biosystems software. The relative transcript expression was calculated by using the 2-ΔΔT.Citation39 Fold changes in the gene expression in comparison with the control were determined and the error was determined by using the standard error of the mean.
Determining reference gene expression stability
To identify stable normalization genes for quantitative RT-PCR assays in each experimental set, the stability of the mRNA expression of each gene was statistically analyzed by using the following reference gene stability analysis software packages: the NormFinder algorithm and the BestKeeper Excel-based tool.Citation40,Citation41 CT values from the ABI ViiA7 real-time PCR system (Applied Biosystems) were converted into relative quantities and imported into the NormFinder Add-in according to the manufacturer's instructions, and the results were shown as the expression stability. Candidate reference genes with the lowest expression stability were considered to be most stable under tested experimental conditions by combining the results from the analysis conducted with the NormFinder and BestKeeper programs.Citation42,Citation43
RESULTS
The de novo assembly of L. santarosai serovar Shermani
Here we present a hybrid approach (Figure ) for high-quality, whole-genome assemblies as performed de novo for L. santarosai serovar Shermani by using second-generation sequencing technology, namely Illumina paired-ends, mate-paired technology and the 454 GS FLX platform, and third-generation sequencing technology, specifically Single Molecule, Real-Time (SMRT) DNA sequencing technology (PacBio RS). We also used high-resolution, whole-genome restriction endonuclease maps of L. santarosai serovar Shermani to confirm the correct placement of the sequence contigs that were generated during the finishing process. The genome sequence of L. santarosai serovar Shermani that was derived from the Illumina paired-end reads data had been previously deposited in DDBJ/EMBL/GenBank.Citation20 Previously generated assemblies using Short Oligonucleotide Analysis Package de novo are comprised of 111 contigs covering 98.81% of the genome and have an N50 value of 97.5 kilobase (kb). To complete the highly repetitive genome, we sequenced genomic DNA from L. santarosai serovar Shermani by using mate-paired sequencing in the Illumina platform, Roche 454 and PacBio RS sequencing technology.
Figure 1 Workflow for the de novo assembly of the L. santarosai serovar Shermani genome. CI, chromosome I; CII, chromosome II; OM, optical mapping.
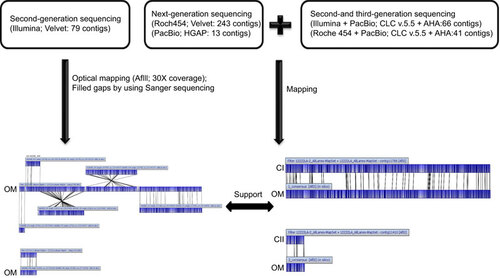
There were three sets of reads used in the assembly approach as follows: (i) Illumina reads, including a Illumina paired-end 500 bp library that generated 7 987 144 reads with approximately 150-fold coverage of the estimated genome size and an Illumina mate-pair 3000 bp library that generated 27 550 738 reads with approximately 550-fold coverage, which were used in the initial sequence assembly to generate scaffolds; (ii) 454-FLX pyrosequencing reads with a total of 597 201 reads (60-fold coverage of the estimated genome size) with average read lengths equal to 410 bp were used for the gap closure of genome sequence assemblies; and (iii) the PacBio RS long-read sequencing platform was generated from a single ∼10-kb SMRT library, which yielded 40 203 continuous long reads with a typical average read length of 3514 bp, and it was used to bridge segments of repetitive regions to form scaffolds. The 10 kb continuous long read data were filtered by read quality (>0.75), resulting in approximately a four-fold coverage of the estimated leptospiral genome size. The sequencing statistics for the L. santarosai serovar Shermani whole-genome mapped reads are given in .
By integrating Illumina paired-end and mate-pair reads with the whole-genome restriction endonuclease maps (AflII; 30-fold coverage) of L. santarosai serovar Shermani, these Illumina reads were assembled de novo by using the Velvet assembler, resulting in six scaffolds with a total size of 3 910 176 bp, and the order of six scaffolds was generated from the high-resolution AflII optical map. We also used Sanger sequencing reads for 131 gaps within and between these scaffolds, and 130 gaps containing 21 gaps with a size of >1 kb were closed. These gaps were closed by direct PCR methods, and the scaffolds were joined together into the first assembly sequences over 3 947 535 bp in length, covering 99.09% of the genome. After filling and closing with unplaced contigs, there was a remaining gap in which the 28 kb fragment was missing relative to the AflII optical map, and hence we suggest that the region may have arisen as highly repeated sequences, or a region of the genome is simply not represented in the read set.
To complete the genome, genomic DNA from L. santarosai serovar Shermani was sequenced and used to generate 454-FLX pyrosequencing reads from 454-FLX pyrosequencing and SMRT sequencing platforms. We tried to assemble contigs from different combination of reads. We used CLC de novo assembly for Illumina and Roche 454 reads with default parameters, which give us 79 and 243 contigs (). These assembled contigs were further assembled with long reads from PacBio. We used two different de novo assembling packages, namely, HGAP and AHA from PacBio SMRT Portal v2.0. The HGAP algorithm generates 13 contigs by using reads from the SMRT sequencing platform only. When applying the AHA to both PacBio and the contigs assembled from Illumina and Roache 454, 66 and 41 contigs were generated, respectively. In addition, we combined de novo contigs generated from an assembly of Illumina and Roche 454 sequence data with contigs assembled from PacBio reads to yield the second assembly sequences, for two nearly finished contigs covering 99% of the genome. In addition, sequences from a gap of 28 kb in size between scaffolding boards were filled by using PacBio RS information.
By mapping the first and second assembly sequences, the genome of L. santarosai serovar Shermani was 100% completed with a total size 3 983 611 bp. To validate the quality of this assembly, all raw reads from second- and third-generation sequencing technology and all Sanger sequence reads were mapped onto the complete genome sequences of L. santarosai serovar Shermani. The L. santarosai serovar Shermani genome was finished without relying on a reference genome, and we addressed three assembly platforms and generated two genome assemblies. The final finished genome sequences were annotated from the Prokaryotic Genomes Automatic Annotation Pipeline (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html). The sequences have been deposited in GenBank under accession numbers CP006694 (chromosome I) and CP006695 (chromosome II).
Table 2 Statistics for de novo assembly of the L. santarosai serovar Shermani strain LT821 (ATCC number 43286) genome.
Features of the L. santarosai serovar Shermani genome
The L. santarosai serovar Shermani genome consists of two circular chromosomes for a total of 3 983 611 bp, with a large one of 3 659 905 bp (chromosome I; CI) and a small one of 323 706 bp (chromosome II; CII), as evidenced by pulsed-field gel electrophoresis analysis (data not shown). Circular representations of both chromosomes are depicted in Figure . Two chromosomes from L. santarosai serovar Shermani have an overall G+C content of 41.82% containing 4079 CDSs with an average length of 830 bp (the largest CDS being 7773 bp), corresponding to a protein-coding content of 92.5%, with 5 genes for ribosomal RNAs (rRNAs) and 37 genes for transfer RNAs ( and Figure ). As previously described, the rRNA genes in L. santarosai serovar Shermani are not organized into operons, as in most other bacteria, but they are scattered over chromosome I.Citation44 L. santarosai serovar Shermani has one rrf gene, two rrl genes and two rrs genes coding for 5S, 23S and 16S rRNAs, respectively. When comparing the complete rrs (16S) sequences for L. santarosai serovar Shermani, L. borgpetersenii and L. interrogans, the shared identity among the sequences is 99% to 98%. The rrf (5S) sequence identity comparing L. santarosai and L. borgpetersenii is 100% and the rrl (23S) is 100%. Based on ribosomal genes, L. santarosai and L. borgpetersenii are closely related, as supported by the whole-genome comparison. In comparison with the previous annotation of the L. santarosai serovar Shermani draft genome by using reciprocal BLASTx searches, it appears that an increase of 198 genes containing two genes for membrane protein, one gene for type IV pilus, one gene for imelysinCitation45 and 182 mostly hypothetical structural genes were identified from the finished genome. Imelysin-like proteins involved in iron uptake are widely distributed in bacteria. In addition, functional annotations of the finished genome sequence for L. santarosai serovar Shermani yielded 2612 hits for the Clusters of Orthologous Groups database.Citation46 Based on the Clusters of Orthologous Group functional classification scheme, genes encoding proteins are involved in function category L (replication, recombination and repairs) and function category D (cell cycle control, cell division and chromosome partitioning) in L. santarosai serovar Shermani compared with L. interrogans, L. borgpetersenii and L. biflexa, which may likely contribute to L. santarosai for the adaptation of the selective pressures and evolutionary developments that allowed its survival in a wide variety of environments such as animal and aquatic environments.
Figure 2 A circular representation of the L. santarosai serovar Shermani genome, with predicted CDSs. (A) Chromosome I; (B) chromosome II. The inner scale is shown in kb. Circles range from 1 (outer circle) to 6 (inner circle). Circles 1 and 3, genes on forward and reverse strands of CDSs; circles 2 and 4, genes on forward and reverse strands of Clusters of Orthologous Group categories; All genes are color-coded according to their functions: red for lipid transport and metabolism (I), lime for carbohydrate metabolism (G), tan for coenzyme transport and metabolism (H), maroon for translation, ribosomal structure and biogenesis (J), blue for cell motility (N), goldenrod for inorganic ion transport and metabolism (P), cyan for post-translation modification, protein turnover and chaperones (O), plum for signal transduction mechanism (T), yellow for secondary metabolites biosynthesis (Q), green for amino acid transport and metabolism (E), olive for energy production and conversion (C), dark khaki for cell division/chromosome partitioning (D), magenta for nucleotide transport and metabolism (F), indigo for transcription (K), purple for replication, recombination and repair (L), dark cyan for cell wall/membrane/envelope biogenesis (M), dull gray for general function prediction only (R), silver for unknown functions (S); circle 5, GC content; circle 6, GC bias ((G-C)/(G.C). This figure was prepared in CGView.
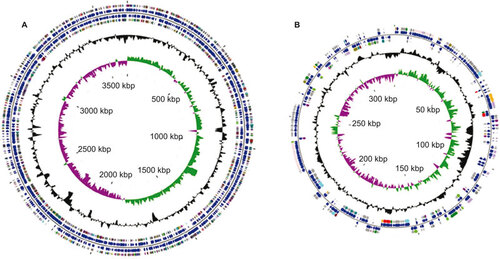
The presence of a large number of mobile genetic elements and clustered regularly interspaced short palindromic repeats (CRISPRs) may be a typical characteristic associated with Leptospira species that have the genome plasticity required for survival both within mammalian hosts and aquatic environments.Citation47 The genome of L. santarosai serovar Shermani has 48 transposases and 5 CRISPR genes homology, indicating that the L. santarosai genomic architecture may have undergone complex genetic alterations and genetic reshuffling during its evolutionary history.
The genome of L. santarosai serovar Shermani was compared with those of previously published pathogenic Leptospira spp. by using a genome alignment with progressive MAUVE.Citation48 A comparison analysis revealed that 11 unique regions, each with sizes greater than 10 kb in size, were found in the complete genome of L. santarosai serovar Shermani and 7 of which were identified as possible genomic islands (GIs) by web-based IslandViewer software.Citation49 In addition, comparative genetic analysis based on reciprocal BLASTp searches revealed that 32 unique CDSs encoding for hypothetical proteins were within these GIs. The Pfam analysis of these unique hypothetical proteins in L. santarosai serovar Shermani indicates that the LSS19962 protein belongs to the peptidase C39-like family. The unique genes in GIs from the L. santarosai serovar Shermani whole genome that are larger than or equal to 150 bp in length are summarized in . Taken together, the whole-genome comparison maps were visualized with CGView Comparison Tool software as shown in Figure .
Figure 3 A circular genome map for L. santarosai serovar Shermani compared with pathogenic Leptospira spp. The sequence similarity detected by BLASTp comparison analysis of chromosome I (A) and chromosome II (B) of pathogenic Leptospira spp. using L. santarosai serovar Shermani as a reference were performed with CGView Comparison Tool software. The circles are colored according to the percent identities of matches (black to light red, 100%–50% identity; blue to light blue, 50%–10% identity; and colorless, 0% identity). From the inner to outer circle on A: GC skew and GC content of L. santarosai serovar Shermani, L. borgpetersenii Hardjo-bovis serovar JB197, serovar L550, L. interrogans Copenhageni serovar Fiocruz L1-130, L. interrogans Lai serovar 56601 and strain IPAV, forward and reverse strand CDSs of L. santarosai serovar Shermani. From the inner to outer circle on B: GC skew and GC content of L. santarosai serovar Shermani, L. interrogans Copenhageni serovar Fiocruz L1-130, L. borgpetersenii Hardjo-bovis serovar JB197, L. interrogans Lai serovar 56601 and strain IPAV, L. borgpetersenii Hardjo-bovis serovar L550, forward and reverse strand CDSs of L. santarosai serovar Shermani.
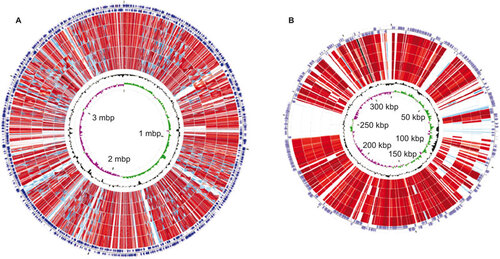
In considering genes that are common to Leptospira species, direct comparisons between the predicted CDSs of L. santarosai and previously sequenced Leptospira species genomes were performed by reciprocal BLASTx searches, and we did not consider predicted CDSs that were less than or equal to 150 bp in length that lacked significant homologs. The result revealed that approximately 126 genes with no hits or non-significant hits in previously sequenced Leptospira species genome are only present in L. santarosai. Of these non-orthologous genes that are unique to L. santarosai serovar Shermani, 124 genes were for hypothetical proteins, and three genes (LSS08219, LSS19048 and LSS21610) were for metallophosphoesterase, peptidase M15A and transposase IS3, respectively. To understand the function of these unique hypothetical proteins in L. santarosai serovar Shermani, a Pfam-based motif/domain analysis was performed as part of this study. Of the 124 unique hypothetical genes, 4 were predicted to be a group II intron, one was predicted to be a lipoprotein and LSS19962 encoded a protein belonging to a peptidase C39-like family. We identified genes that were unique to L. santarosai serovar Shermani in this study and further examined these species-specific genes for their role in the pathogenesis and clinical diagnosis benefits that should be conducted as necessary.
Table 3 Unique regions belonging to GIs and containing unique genes in L. santorosai serovar Shermani
Analyzing the gene sequences of L. santarosai serovar Shermani strain CCF
To investigate sequence similarities between L. santarosai serovar Shermani strain LT821 and strain CCF, we examined the conservation of genes in an isolate of L. santarosai serovar Shermani genome and a sequence alignment analysis by using a VectorNTI tool.Citation50 L. santarosai serovar Shermani strain CCF, a clinically important leptospire that was confirmed by microscope agglutination test, was isolated from a Taiwanese patient with leptospirosis who had acute tubulointerstitial nephritis.Citation29 According to the information from whole-genome sequences of the L. santarosai serovar Shermani strain LT821, we selected 30 CDSs with 17 for hypothetical genes and seven for lipoprotein to elucidate the presence or absence of these genes in the L. santarosai serovar Shermani strain CCF genome sequences.
Of these hypothetical genes, nine genes (LSS19413, LSS22130, LSS16416, LSS16441, LSS23260, LSS20184, LSS05845, LSS19962 and LSS20521) were unique in L. santarosai serovar Shermani strain LT821 and four genes (LSS22130, LSS16416, LSS16441 and LSS19962) were located in predicated GI regions. The primers used for gene-specific amplification are shown in Supplementary Table S1. PCR products were separated on agarose gels, and the migration of the corresponding products from L. santarosai serovar Shermani strain LT821 is shown to compare the PCR product from L. santarosai serovar Shermani strain CCF (Supplementary Figure S1). The successfully amplified products were recovered by gel extraction and further verified by sequencing at the DNA Sequencing Core Laboratory (Chang Gung Memorial Hospital, Linkou, Taiwan). This analysis revealed the presence of these gene elements and the same size PCR products in each primer set between L. santarosai serovar Shermani strain LT821 and strain CCF. Under the standard concentration of genomic DNA (500 ng), PCR product results differed slightly in brightness as shown for LSS16416, LSS0621, LSS05845, LSS07729, LSS14677, LSS13644, LSS13769, LSS23260, LSS15341, LSS11940 and LSS12614, explaining the number of gene variations between L. santarosai serovar Shermani strain LT821 and strain CCF. In addition, sequence similarity analysis revealed that these PCR products from two different strains of L. santarosai serovar Shermani exhibited no differences, suggesting a strong identity within L. santarosai serovar Shermani. These data indicated that there is strong sequence conservation between L. santarosai serovar Shermani strain LT821 and strain CCF.
A comparative analysis of differential leptospiral gene expression for L. santarosai serovar Shermani- and L. interrogans serovar Copenhageni-infected HK-2 cells
L. santarosai serovar Shermani is the most frequently isolated serovar, and it causes both renal and systemic infections in Taiwan. From a clinical presentation perspective, L. santarosai serovar Shermani causes tubulointerstitial nephritis similar to that of other strains that induce nephritis, for example L. interrogans serovar Copenhageni. Nevertheless, it is interesting to note that leptospiral morphological differences exist between L. santarosai- and L. interrogans-infected HK-2 cells. After 4 h of infection at an multiplicity of infection of 100, L. interrogans serovar Copenhageni had a typical spiral shape. However, L. santarosai serovar Shermani tended to aggregate in culture conditions that were observed by using immunohistochemistry (with a polyclonal anti-leptospiral LipL32 antibody) (data not shown). L. santarosai serovar Shermani had a morphologically distinct form in an infectious condition with HK-2 cells, suggesting differential gene expressions for L. santarosai serovar Shermani- and L. interrogans serovar Copenhageni-infected HK-2 cells. According to data collected from transcriptomic approaches (data not shown), 12 gene targets containing 5 for hypothetical genes, 4 for lipoproteins and 1 for motility were selected to analyze in this study (Supplementary Table S1 and Supplementary Table S2). By using BLAST to find homologous genes between L. santarosai serovar Shermani and L. interrogans serovar Copenhageni genomes, the genes selected for this study are listed in . The co-cultivation of pathogenic Leptospira spp. with HK-2 cells was used as an infection model to study leptospiral gene expressions. After infecting with L. santarosai serovar Shermani and L. interrogans serovar Copenhageni for 4 h in the cell medium without antibiotics and then harvesting, the bacteria were subjected to RNA extraction, cDNA synthesis and quantitative RT-PCR analysis.
In our cell-based-infection model study, the 16S rRNA gene was not a suitable potential reference gene/housekeeping gene, as evidence by the threshold cycle value of the 16S rRNA gene detected in a sample harvested from HK-2 cells without any bacteria. A combination of Normfinder and BestKeeper analyses were used to choose the best housekeeping gene. These tested genes were analyzed for the most stable control gene with the lowest expression stability, as defined as an internal control that was considered to be most stable under the tested experimental conditions. The stability value was 0.135 for the best combination of two gene targets, namely, LSS16476 and LSS08624, the most stable genes under our experimental conditions in which the samples (with a total of 18 genes for analysis) were harvested from HK-2 cells infected with L. santarosai serovar Shermani. In another, the stability value was 0.361 for the best gene target, or LIC12263, which was the most stable gene under our experimental condition from the samples (with a total of 16 genes for analysis) as harvested from HK-2 cells infected with L. interrogans serovar Copenhageni.
In the L. santarosai serovar Shermani-HK-2 cell infection model, LSS21190 encoding imelysin is downregulated (fold change <0.5) after infection for 4 h. We show that LSS14677 encoding OmpL37, LSS01089 encoding hypothetical protein, LSS19962 encoding a hypothetical protein with a C39-like domain, LSS16716 encoding FlaB, LSS15341 encoding LipL21 and LSS00320 encoding LipL36 are upregulated (fold change >2) after infection for 4 h. In addition, the transcripts of LSS08269 encoding a hypothetical protein with an ankyrin repeat involved in function category R (general functional prediction only) and LSS16296 encoding Lsa24 were not detected, most likely reflecting a very low expression level (Figure ).
Figure 4 A comparative analysis of differential leptospiral gene expressions in L. santarosai serovar Shermani-infected HK-2 cells (A) and L. interrogans serovar Copenhageni-infected HK-2 cells (B). Data are represented as the means±SD of three independent experiments.
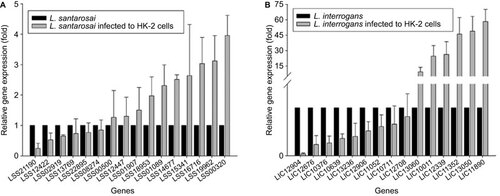
In L. interrogans serovar Copenhageni-HK-2 cell infection model, LIC12676 and LIC10376 encoding hypothetical protein, and LIC12904 encoding a von Willebrand factor A-domain-containing protein are downregulated (fold change <0.5) after infection for 4 h. The results show that LIC11352 encoding LipL32, LIC13050 encoding a hypothetical protein, LIC11890 encoding FlaB, LIC10011 encoding LipL21, LIC13060 encoding LipL36 and a LIC12339 gene belonging to a paralogous (PF07598) gene family are upregulated (fold change >2) after infection for 4 h (Figure ). The most interesting result was the dramatic upregulation (almost >10-fold) of these lipoproteins in L. interrogans serovar Copenhageni upon interaction with HK-2 cells.
In this study, lipoprotein gene expression in both L. santarosai serovar Shermani and L. interrogans serovar Copenhageni were upregulated upon interaction with HK-2 cells in comparison with the culture medium controls alone. Our results indicated that the interaction with human renal proximal tubular cells for 4 h was an important factor in triggering the differential expression of lipoprotein in pathogenic Leptospira. spp. It can be hypothesized that lipoprotein is required for attachment only during the early stages of the infection through human renal proximal tubular cells.
Table 4 Genes for leptospiral gene expression analysis in cell-based infection models.
DISCUSSION
Here we present the first report of the sequencing and de novo assembly of a Leptospira santarosai serovar Shermani genome by using a hybrid sequencing strategy in which we combined high-accuracy, short-read data from second-generation sequencing technologies with long-read PacBio data and data interpretation that was performed in a renal tubular cell-based infection model that may elucidate leptospira pathogenesis in kidneys. L. santarosai belongs to a less-studied pathogenic species of leptospires. We completed the whole-genome sequences of L. santarosai and further compared it with other recently sequenced Leptospira spp. genomes. To investigate the virulence factor of L. santarosai serovar Shermani and to improve the available genome sequences for comparative analysis, we used L. santarosai serovar Shermani (ATCC number 43286) for complete genome sequencing. Because of sequencing biases and high repetitive genomic features of Leptospira spp. that make certain regions difficult or impossible to assemble, we combined second-generation sequencing, namely Illumina paired-end, mate-paired sequencing and the 454 GS FLX platform, and third-generation sequencing technology, specifically PacBio RS SMRT DNA sequencing technology. Furthermore, the high-resolution optical mapping platform and Sanger-based manual finishing processes were used to complete the genome sequences. Our hybrid assembly protocol could resolve complex repeat-rich segments of the Leptospira spp. genome.
In our previous studies, Illumina sequencing data with shorter reads (<150 bp) and a De Bruijn graph-based assembler (Velvet and CLC de novo assembler) were used for the de novo assembly of the draft leptospiral genome sequences, which consisted of a total of 110 contigs. Notwithstanding the announcement of the Leptospira genome draft sequence, we are mindful that it contains gaps and nucleotide errors. Moreover, fully sequenced bacterial genomes are superior to draft whole genomes because they provide the only accurate reference for interpreting transcriptomes. To address this issue, we adopted Roche 454 pyrosequencing to generate hundreds of thousands of long reads (<450 bp). However, our results show that sole reliance on second-generation sequencing technologies cannot usually produce a complete Leptospira genome with highly repeated regions greater than 1000 bp in length. Therefore, we then included the PacBio RS sequencing platform, which is a third generation sequencing technology and can produce reads longer than large repeats within the genome. Because PacBio reads are known for their relatively higher error rate, the methods HGAP and AHA were further adopted. Both HGAP and AHA are specifically developed for PacBio reads and can fix potential sequencing error. To validate the quality of this assembly in our study, all raw reads from NGS platforms and all Sanger sequence reads were mapped onto the finished genome sequences, revealing that this assembly genome were covered by these reads, but not all raw reads were mapped onto this assembly genome. In addition, we also designed primers and checked conflict regions (coverage <20) with Sanger sequencing.
CRISPRs, which are putatively antiviral elements for host defense mechanisms against bacteriophage predation, are a class of repetitive DNA elements that are propagated via horizontal gene transfer in prokaryotes.Citation51 The CRISPR elements have been detected in L. santarosai in addition to L. interrogans, but not in L. borgpetersenii and L. biflexa. The presence of CRISPR elements in the L. santarosai genome suggests the presence of genetic alterations and exchanges as a result of bacteriophage infection during evolutionary pressure. In addition, we cannot find putative type II toxin–antitoxin systems that contribute to the stable maintenance and dissemination of plasmids and GIs in L. santarosai, suggesting that mobile genetic elements encoding toxin–antitoxin systems are lost during cell division or simply cannot be identified by our annotated method.
Mobile DNA elements including prophages, transposons and insertion sequence elements abound in Leptospira spp. A comparative genome analysis in Leptospira spp. showed that the number of transposases vary among the species, ranging from 26 in L. interrogans and 48 in L. santarosai to 241 in L. borgpetersenii, suggesting genome plasticity in Leptospira species. In addition, a group II intron can be transferred between bacteria on conjugative elements and move from site to site within a bacterium by retrotransposition, and it was previously was identified in L. borgpetersenii to provide evidence for lateral transfer in Leptospira.Citation13 Interestingly, unique genes predicted as a group II intron are found in the L. santarosai genome. According to this study, we infer that L. santarosai unique regions with acquisition through horizontal gene transfer may contain genes specifying virulence traits in addition to genes that may enhance fitness in a specific environmental niche. Moreover, L. santarosai and L. borgpetersenii genomes are similar in size (), but the gene density in L. santarosai is much higher, most likely reflecting the greater genetic information required for L. santarosai to survive within both mammalian hosts and aquatic environments.
GIs, which provide evidence of lateral gene transfer and contain genes for functions involved in symbiosis or pathogenesis, may introduce virulence factors into a new host genome.Citation52 Thirteen predicted GIs in L. santarosai ranging in sizes above 10 kb were identified by using the online tool IslandViewer (http://www.pathogenomics.sfu.ca/islandviewer),Citation49 which integrates three different GI prediction methods (IslandPick, IslandPath-DIMOB and SIGI-HMM). Of these GIs, there are seven GIs located within unique regions in the L. santarosai serovar Shermani genome. In addition, the sequences of a 28 kb gap in the size between scaffolding boards were filled by using PacBio RS information during assembly processes. Interestingly, a unique 28-kb region in L. santarosai had been predicted as GI-containing genes that encode transposases, lipoproteins and hypothetical proteins. Furthermore, it revealed the presence of a unique 28-kb region in L. santarosai serovar Shermani strain LT821, and in an isolate from a Taiwanese patient called strain CCF, but it is absent in other pathogenic Leptospira spp. In this region, LSS19962-encoding hypothetical proteins belonging to a C39-like family were present in L. santarosai, but they were absent in other pathogenic Leptospira spp. LSS19962. This gene is unique, and it was upregulated in the L. santarosai serovar Shermani-HK-2 cell infection model, further suggesting that its functions might be related to adherence or virulence. Genes unique to L. santarosai are likely to be necessary for aiding infection (pathogenesis).
Pathogenic L. santarosai serovars have been classified into group I pathogens, which cause disease in humans with varying degrees of severity.Citation53 A chance discovery and experimental evidence showed that virulence-associated genes belonging to a PF07598 gene family present in group I pathogens, but not in group II pathogens and saprophytic species, are highly upregulated during infection relating to kidney colonization. There are two PF07598 paralogs called LSS14871 and LSS03359 that encode hypothetic proteins in the finished whole-genome sequence of L. santarosai serovar Shermani LT821.
In this study, the new whole-genome sequence information was useful to develop PCR-based gene markers. A PCR using primers designed on the basis of L. santarosai serovar Shermani strain LT821 DNA sequences was used to investigate the presence of these genes in the L. santarosai serovar Shermani strain CCF genome sequences, a clinical strain. These results provided evidence of sequence conservation between L. santarosai serovar Shermani strain LT821 and strain CCF, in addition to having slight gene number variations. Ideally, the availability of the whole-genome sequence of L. santarosai serovar Shermani might have an impact on clinical diagnostic applications.
Next-generation sequencing-based transcriptome responses to L. santarosai serovar Shermani strain LT821 infection in HK-2 cells were performed by using sequencing by oligonucleotide ligation and detection technology (data not shown). After aligning RNA-seq reads to the L. santarosai serovar Shermani strain LT821 genome, the digital expression levels (RPKM, reads per kilobase of exon model per million mapped reads) of each annotated leptospiral genes were calculated. By using a RPKM value of >1000, there are 33 leptospiral genes expressed during the cocultivation of L. santarosai serovar Shermani strain LT821 with HK-2 cells for a 4 h incubation. However, the PF07598 paralogs LSS14871 and LSS03359 had RPKM values of <10, indicating that there was lower gene expression under the HK-2 cell-based infection condition. An analysis of RNA-seq data for L. santarosai serovar Shermani-infected HK-2 cells revealed 207 transcripts with differential expression (with a fold change of ≥4) between groups at different time points post-infection (2 h vs. 4 h). The complete RNA-seq data will be reported in a separate manuscript that is in preparation. According to data from genome and RNA-seq analysis, species-specific genes and lipoprotein genes were chosen for a comparative analysis of differential leptospiral gene expressions in L. santarosai serovar Shermani and L. interrogans serovar Copenhageni-infected HK-2 cells. Interestingly, we found that the expression of several lipoprotein genes (e.g., lipL32, 21 and 36) was more upregulated in an L. interrogans serovar Copenhageni infection in HK-2 cells than in an L. santarosai serovar Shermani strain LT821 infection in HK-2 cells. In addition, our results revealed the transcripts of LSS01089 encoding hypothetical proteins, LSS15341 (for LipL21), LSS00320 (LipL36) and LSS16716 (for FlaB) were upregulated in L. santarosai serovar Shermani strain LT821 infection in HK-2 cells, which was consistent with previous information from the transcriptome study. Taken together, we suggest that the interaction with HK-2 cells may be an important factor in triggering the differential expression of major OMPs or lipoproteins in Leptospira spp. This finding was in contrast to previous evidence showing that the downregulation of this group of major OMPs or lipoproteins was likely an immune evasion mechanism of L. interrogans. In our infection models, this finding supports the hypothesis that OMPs and lipoproteins may be expressed during leptospiral infection and facilitates attachment to the kidney. The overall results derived from the combined computational genome analysis and correlation with the available experimental evidence may be useful in the discovery of novel genes and for understanding the pathogenicity of Leptospira.
Supplementary Figure S1
Download PDF (138.4 KB)Supplementary Table S1
Download MS Word (35.1 KB)Supplementary Table S2
Download MS Word (29.6 KB)This work was supported by Chang Gung Memorial Hospital Grants CMRPG390692 and National Science Council Grants NSC100-2314-B-182-031-MY3. We gratefully acknowledge Dr Chih-Peng Lin (Department of Bioinformatics, Yourgene Bioscience, Xinbei, Taiwan) for his generous assistance with genome assembly. We thank the DNA Sequencing Core Laboratory (Chang Gung Memorial Hospital, Taoyuan, Taiwan) for the Sanger sequencing.
Supplementary Information for this article can be found on Emerging Microbes and Infections' website (http://www.nature.com/emi/).
- Adler B, de la Pena Moctezuma A.Leptospira and leptospirosis. Vet Microbiol2010;140: 287–296.
- Evangelista KV, Coburn J.Leptospira as an emerging pathogen: a review of its biology, pathogenesis and host immune responses. Future Microbiol2010;5: 1413–1425.
- Bharti AR, Nally JE, Ricaldi JN et al.Leptospirosis: a zoonotic disease of global importance. Lancet Infect Dis2003;3: 757–771.
- Lin PC, Chi CY, Ho MW, Chen CM, Ho CM, Wang JH.Demographic and clinical features of leptospirosis: three-year experience in central Taiwan. J Microbiol Immunol Infect2008;41: 145–150.
- Yang CW.Leptospirosis in Taiwan—an underestimated infectious disease. Chang Gung Med J2007;30: 109–115.
- Yang CW, Pan MJ, Wu MS et al.Leptospirosis: an ignored cause of acute renal failure in Taiwan. Am J Kidney Dis1997;30: 840–845.
- Yang CW, Wu MS, Pan MJ.Leptospirosis renal disease. Nephrol Dial Transplant2001;16(Suppl 5): 73–77.
- Subrahmanian PS, Abraham G, Thirumurthi K, Mathew M, Reddy YN.Reversible acute kidney injury due to bilateral papillary necrosis in a patient with leptospirosis and diabetes mellitus. Indian J Nephrol2012;22: 392–394.
- Ko AI, Goarant C, Picardeau M.Leptospira: the dawn of the molecular genetics era for an emerging zoonotic pathogen. Nat Rev Microbiol2009;7: 736–747.
- Cerqueira GM, Picardeau M.A century of Leptospira strain typing. Infect Genet Evol2009;9: 760–768.
- Xue F, Yan J, Picardeau M.Evolution and pathogenesis of Leptospira spp.: lessons learned from the genomes. Microbes Infect2009;11: 328–333.
- Nascimento AL, Verjovski-Almeida S, van Sluys MA et al.Genome features of Leptospira interrogans serovar Copenhageni. Braz J Med Biol Res2004;37: 459–477.
- Bulach DM, Zuerner RL, Wilson P et al.Genome reduction in Leptospira borgpetersenii reflects limited transmission potential. Proc Natl Acad Sci USA2006;103: 14560–14565.
- Picardeau M, Bulach DM, Bouchier C et al.Genome sequence of the saprophyte Leptospira biflexa provides insights into the evolution of Leptospira and the pathogenesis of leptospirosis. PLoS One2008;3: e1607.
- Adler B, Lo M, Seemann T, Murray GL.Pathogenesis of leptospirosis: the influence of genomics. Vet Microbiol2011;153: 73–81.
- Ricaldi JN, Fouts DE, Selengut JD et al.Whole genome analysis of Leptospira licerasiae provides insight into leptospiral evolution and pathogenicity. PLoS Negl Trop Dis2012;6: e1853.
- Ren SX, Fu G, Jiang XG et al.Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature2003;422: 888–893.
- Zhong Y, Chang X, Cao XJ et al.Comparative proteogenomic analysis of the Leptospira interrogans virulence-attenuated strain IPAV against the pathogenic strain 56601. Cell Res2011;21: 1210–1229.
- Nascimento AL, Ko AI, Martins EA et al.Comparative genomics of two Leptospira interrogans serovars reveals novel insights into physiology and pathogenesis. J Bacteriol2004;186: 2164–2172.
- Chou LF, Chen YT, Lu CW et al.Sequence of Leptospira santarosai serovar Shermani genome and prediction of virulence-associated genes. Gene2012;511: 364–370.
- Wilson MR, Naccache SN, Samayoa E et al.Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl J Med2014;370: 2408–2417.
- Hartwig DD, Seixas FK, Cerqueira GM, McBride AJ, Dellagostin OA.Characterization of the immunogenic and antigenic potential of putative lipoproteins from Leptospira interrogans. Curr Microbiol2011;62: 1337–1341.
- Haake DA, Matsunaga J.Characterization of the leptospiral outer membrane and description of three novel leptospiral membrane proteins. Infect Immun2002;70: 4936–4945.
- Yang CW, Hung CC, Wu MS et al.Toll-like receptor 2 mediates early inflammation by leptospiral outer membrane proteins in proximal tubule cells. Kidney Int2006;69: 815–822.
- Hung CC, Chang CT, Chen KH et al.Upregulation of chemokine CXCL1/KC by leptospiral membrane lipoprotein preparation in renal tubule epithelial cells. Kidney Int2006;69: 1814–1822.
- Yang CW, Wu MS, Pan MJ, Hsieh WJ, Vandewalle A, Huang CC.The Leptospira outer membrane protein LipL32 induces tubulointerstitial nephritis-mediated gene expression in mouse proximal tubule cells. J Am Soc Nephrol2002;13: 2037–2045.
- Hung CC, Chang CT, Tian YC et al.Leptospiral membrane proteins stimulate pro-inflammatory chemokines secretion by renal tubule epithelial cells through Toll-like receptor 2 and p38 mitogen activated protein kinase. Nephrol Dial Transplant2006;21: 898–910.
- Barnett JK, Barnett D, Bolin CA et al.Expression and distribution of leptospiral outer membrane components during renal infection of hamsters. Infect Immun1999;67: 853–861.
- Hsieh WJ, Chang YF, Chen CS, Pan MJ.Omp52 is a growth-phase-regulated outer membrane protein of Leptospira santarosai serovar Shermani. FEMS Microbiol Lett2005;243: 339–345.
- Minas K, McEwan NR, Newbold CJ, Scott KP.Optimization of a high-throughput CTAB-based protocol for the extraction of qPCR-grade DNA from rumen fluid, plant and bacterial pure cultures. FEMS Microbiol Lett2011;325: 162–169.
- Zerbino DR, Birney E.Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res2008;18: 821–829.
- Chin CS, Alexander DH, Marks P et al.Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods2013;10: 563–569.
- Bashir A, Klammer AA, Robins WP et al.A hybrid approach for the automated finishing of bacterial genomes. Nat Biotechnol2012;30: 701–707.
- Zhou S, Kile A, Bechner M et al.Single-molecule approach to bacterial genomic comparisons via optical mapping. J Bacteriol2004;186: 7773–7782.
- Stothard P, Wishart DS.Circular genome visualization and exploration using CGView. Bioinformatics2005;21: 537–539.
- Grant JR, Stothard P.The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res2008;36( Web Server issue): W181–W184.
- Grant JR, Arantes AS, Stothard P.Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics2012;13: 202.
- Finn RD, Mistry J, Schuster-Bockler B et al.Pfam: clans, web tools and services. Nucleic Acids Res2006;34( Database issue): D247–D251.
- Livak KJ, Schmittgen TD.Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods2001;25: 402–408.
- Andersen CL, Jensen JL, Orntoft TF.Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res2004;64: 5245–5250.
- Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP.Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol Lett2004;26: 509–515.
- Carrillo-Casas EM, Hernandez-Castro R, Suarez-Guemes F, de la Pena-Moctezuma A.Selection of the internal control gene for real-time quantitative rt-PCR assays in temperature treated Leptospira. Curr Microbiol2008;56: 539–546.
- Latham GJ.Normalization of microRNA quantitative RT-PCR data in reduced scale experimental designs. Methods Mol Biol2010;667: 19–31.
- Fukunaga M, Mifuchi I.Unique organization of Leptospira interrogans rRNA genes. J Bacteriol1989;171: 5763–5767.
- Xu Q, Rawlings ND, Farr CL et al.Structural and sequence analysis of imelysin-like proteins implicated in bacterial iron uptake. PLoS One2011;6: e21875.
- Wolf YI, Makarova KS, Yutin N, Koonin EV.Updated clusters of orthologous genes for Archaea: a complex ancestor of the Archaea and the byways of horizontal gene transfer. Biol Direct2012;7: 46.
- Lehmann J, Matthias M, Vinetz J, Fouts D.Leptospiral pathogenomics. Pathogens2014;3: 280–308.
- Darling AC, Mau B, Blattner FR, Perna NT.Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res2004;14: 1394–1403.
- Langille MG, Brinkman FS.IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics2009;25: 664–665.
- Lu G, Moriyama EN.Vector NTI, a balanced all-in-one sequence analysis suite. Brief Bioinform2004;5: 378–388.
- Al-Attar S, Westra ER, van der Oost J, Brouns SJ.Clustered regularly interspaced short palindromic repeats (CRISPRs): the hallmark of an ingenious antiviral defense mechanism in prokaryotes. Biol Chem2011;392: 277–289.
- Juhas M, van der MeerJR, Gaillard M, Harding RM, Hood DW, Crook DW.Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol Rev2009;33: 376–393.
- Pallen MJ, Wren BW.Bacterial pathogenomics. Nature2007;449: 835–842.