
Abstract
Avian influenza subtype H9N2 is endemic in many bird species in Asia and the Middle East and has contributed to the genesis of H5N1, H7N9 and H10N8, which are potential pandemic threats. H9N2 viruses that have spread to Bangladesh have acquired multiple gene segments from highly pathogenic (HP) H7N3 viruses that are presumably in Pakistan and currently cocirculate with HP H5N1. However, the source and geographic origin of these H9N2 viruses are not clear. We characterized the complete genetic sequences of 37 Bangladeshi H9N2 viruses isolated in 2011–2013 and investigated their inter- and intrasubtypic genetic diversities by tracing their genesis in relationship to other H9N2 viruses isolated from neighboring countries. H9N2 viruses in Bangladesh are homogenous with several mammalian host-specific markers and are a new H9N2 sublineage wherein the hemagglutinin (HA) gene is derived from an Iranian H9N2 lineage (Mideast_B Iran), the neuraminidase (NA) and polymerase basic 2 (PB2) genes are from Dubai H9N2 (Mideast_C Dubai), and the non-structural protein (NS), nucleoprotein (NP), matrix protein (MP), polymerase acidic (PA) and polymerase basic 1 (PB1) genes are from HP H7N3 originating from Pakistan. Different H9N2 genotypes that were replaced in 2006 and 2009 by other reassortants have been detected in Bangladesh. Phylogenetic and molecular analyses suggest that the current genotype descended from the prototypical H9N2 lineage (G1), which circulated in poultry in China during the late 1990s and came to Bangladesh via the poultry trade within the Middle East, and that this genotype subsequently reassorted with H7N3 and H9N2 lineages from Pakistan and spread throughout India. Thus, continual surveillance of Bangladeshi HP H5N1, H7N3 and H9N2 is warranted to identify further evolution and adaptation to humans.
Introduction
Since 2009, H9N2 has been the predominant avian influenza virus (AIV) subtype, followed by the highly pathogenic avian influenza (HPAI) H5N1 subtype, isolated from domestic land-based poultry in Bangladesh.Citation1,Citation2,Citation3,Citation4 Molecular characterization of the Bangladeshi H9N2 subtype isolated between 2009 and 2011 reveals that it has acquired numerous molecular markers that influence mammalian host specificity, and three of its eight genes are similar to those of HP H7N3 influenza viruses isolated from Pakistan.Citation4,Citation5 The first human H9N2 infection in Bangladesh was identified in 2011, and the virus subtype was antigenically and molecularly closely related to the then-circulating avian H9N2 viruses.Citation4,Citation6 Despite evidence supporting that H9N2 can infect humans, the importance of H9N2 has been overshadowed by HP H5N1. However, the recent emergence of two avian influenza subtypes, H7N9 and H10N8, which have infected humans in China, highlights the importance of H9N2 because the internal genes of both subtypes originate from H9N2.Citation7,Citation8,Citation9,Citation10
Since the early 1990s, H9N2 viruses in land-based poultry have evolved from a less to a more diverse genotype through the acquisition of gene segments from other viruses. Historically, on the basis of their genetic and antigenic properties, H9N2 viruses circulating in Asia have been classified into three genotypes: A/Quail/Hong Kong/G1/97-like (G1-like); the Y280 lineage, represented by A/Chicken/Hong Kong/Y280/97-like (Y280-like); and the Korean lineage, represented by A/Chicken/Korea/38349-p96323/96 (Korean-like).Citation11,Citation12,Citation13,Citation14 A/Quail/Hong Kong/G1/97, the G1 prototype virus, was the donor of internal genes for HP H5N1 viruses that infected humans in Hong Kong in 1997,Citation14 and it is widely circulating in quails in southeastern China.Citation15
Since 1997, H9N2 viruses descended from G1 lineages have been isolated from influenza outbreaks in the Middle East, Pakistan and India.Citation16,Citation17,Citation18 Between 2005 and 2008, a new H9N2 genotype, represented by A/Chicken/Pakistan/UDL-01/06 (H9N2), which did not cluster in an established H9N2 lineage (hemagglutinin (HA) lineage), was identified in Pakistan's poultry population, and this genotype replaced the G1 lineage.Citation19 A/Chicken/Pakistan/UDL-01/06 (H9N2) resulted from the reassortment of genes from H9N2 (G1-like), HPAI H5N1 (clade 2.2), and HPAI H7N3 (non-structural protein, NS). During the same period, a single H9N2 isolate (A/Chicken/Bangladesh/VP01/2006) having the same genetic backbone as A/Chicken/Pakistan/UDL-01/06 (H9N2) was isolated in Bangladesh. A/Chicken/Bangladesh/VP01/2006 reassorted further with HP H7N3, resulting in a newer genotype that possessed all of the internal genes of HP H7N3.Citation5 Recent studies on H9N2 viruses from Bangladesh suggest that H9N2 viruses are related to the G1 clade, but are very distinct from them and very similar to H9N2 viruses isolated from Pakistan and the Middle East.Citation4
Genetic analyses of the H9N2 viruses isolated from India during 2003 show that they group with the G1-like lineage.Citation20 Phylogenetic analyses identified four distinct H9N2 subgroups in viruses circulating in the Middle East (A, B, C and D), each of which has undergone widespread inter- and intrasubtype reassortments.Citation16 H9N2 viruses isolated from Israel, Jordan and Saudi Arabia were in group A; viruses isolated from Pakistan and Afghanistan as well as viruses isolated from Iran in 2003–2009 were in group B; and viruses isolated from the United Arab Emirates (UAE) in 2000–2002 and from Iran in 1998–2007 were in groups C and D, respectively.Citation16 Recently, on the basis of a revised clade nomenclature proposal, H9 HA genes have been classified into 23 distinct clades and five clade-specific outliers.Citation17
After their isolation in 1997 in Hong Kong, H9N2 viruses related to the G1 clade were introduced into chickens, quails and other land-based poultry in Pakistan, the Middle East and Bangladesh. However, the genesis and evolution of avian H9N2 viruses in Bangladesh have not been determined, even though some molecular epidemiology studies of H9N2 isolated from Bangladesh have been reported.Citation2,Citation3,Citation4,Citation5 Studying the genesis of avian H9N2 viruses in Bangladesh is important because the genotypes of Bangladeshi H9N2 viruses are very similar to those of Pakistani H9N2 viruses, which are G1/H7N3 reassortants. Additionally, there are no reports of H7N3 outbreaks in Bangladesh, suggesting that this genesis did not occur in this country. The question of how the G1 lineage of H9N2 viruses was introduced in the Middle East and Pakistan remains unanswered because there is no known direct migratory flyway between China and the Middle East for the introduction of H9N2 viruses through wild birds.Citation21 Therefore, it is important to resolve the genesis of Bangladeshi H9N2 viruses and to explore the role of various poultry types in the ecology and distribution of these viruses.
We generated and characterized the complete genetic sequences of 37 H9N2 viruses isolated from 2011 to 2013 and studied the evolutionary history of Bangladeshi H9N2 viruses by reviewing past and currently circulating H9N2 viruses from neighboring countries. We report the genesis of the currently circulating H9N2 genotype in Bangladesh and the evolution of a new H9N2 sublineage through mutations and reassortment.
MATERIALS AND METHODS
Sample collection and screening
The epidemiological surveillance of avian influenza in Bangladesh was begun in November 2008 and is ongoing. Biological sampling was conducted monthly throughout the year, and samples were collected from live-bird markets (LBMs), farms and wetlands, as previously described.Citation3 From August 2010 to December 2013, 12 640 swab samples were obtained from oropharyngeal, cloacal, fecal and water samples, and during this period, an average of 370 samples were collected each month. However, because several locations had no prevalence of AIVs during that period, the sample number was reduced to 200 in 2013, and collection was refocused to LBMs and to the poultry farms that supplied the markets.
Virus isolation and screening
Oropharyngeal, cloacal, and environmental samples were processed and screened for AIVs using real-time reverse transcription-polymerase chain reaction (rRT-PCR) and influenza A-specific primers and probes, as previously described.Citation3,Citation4 During the 2010–2012 surveillance periods, all rRT-PCR-positive samples were inoculated into embryonated eggs to test for the presence or absence of AIVs. However, in 2013, this strategy was modified: all H5-positive rRT-PCR samples and 10% of the non-H5 influenza A-positive samples were inoculated into embryonated eggs. Viral RNA extraction, subtyping, and sequencing were performed as previously described.Citation3,Citation4
Genetic and phylogenetic analyses
Viral RNA preparation, sequencing, and sequence compilation were performed as previously described.Citation4,Citation22 Alignments of H9N2 sequences that were representative of the location, time, species and sample types from which they were isolated was performed using the CLUSTALW module available within the Bioedit program package V 7.2.3.Citation23 Phylogenetic analyses were carried out as previously described.Citation4
The molecular characteristics of 37 H9N2 AIVs were analyzed as previously described.Citation4 The prediction for potential glycosylation sites in the HA gene was made using the biological sequence analysis server 1.0 available at http://www.cbs.dtu.dk/services/NetNgly. The consensus sequence and the nucleotide percent identity of each influenza gene were calculated using the MegAlign (Lasergene; DNAStar, Inc., Madison, Wisconsin, USA.) software, and the heat map was constructed using Matrix2png.Citation24
RESULTS
Isolation of AIVs
Of the 12 640 collected swab samples, 783 (5.6%) were positive for AIVs by PCR screening and virus isolation. The isolation of AIV was consistently higher in chicken for each year of sampling (Figure ) than for other species such as quails and ducks. Among the collected host species samples, 8.6% of the chicken and 8.5% of the quail samples tested positive for AIV, whereas 1.68% of the duck samples tested positive for AIV. The isolation rate for AIV was slightly lower in chickens during 2011 than during 2010 and 2012 (Figure ); however, the overall isolation rate has been steady since 2010. In 2013, because of the high number of AIV isolations, AIV isolation was performed only for 10% of the non-H5-PCR-positive samples; therefore, the isolation rate shown in Figure for 2013 does not reflect the true isolation rate. However, the isolation rate of AIV steadily increased from 2010 to 2013. In general, virus detection was conducted year-round, with occasional spikes observed most notably during the winter months of 2012 (Figure ). The most commonly isolated subtype was H9N2, followed by H5N1. Almost 10% of the samples were commixtures predominantly consisting of H9N2, H5N1 and Newcastle disease virus. The commixtures from samples obtained from chickens and quails were identified by PCR (Supplementary Table S1).
Figure 1 Annual isolation of all avian influenza subtypes in poultry at different live-bird markets and farms in Bangladesh from August 2010 to December 2013. The total number of samples collected each year were 1198 (2010), 4897 (2011), 3952 (2012) and 2593 (2013).
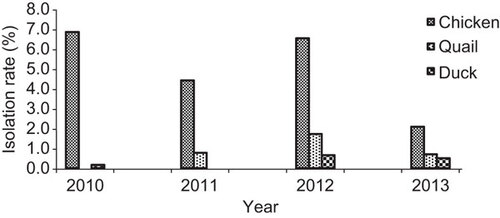
Figure 2 Monthly isolation of all H9N2 and H5N1 subtypes in poultry at different live-bird markets and farms in Bangladesh from August 2010 to December 2013.
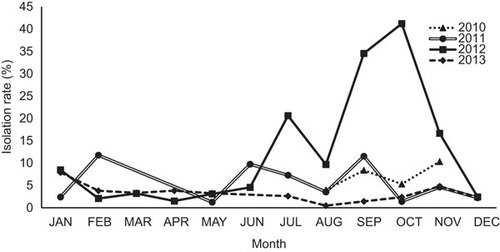
Figure 3 Nucleotide identity matrix of individual genes from Bangladeshi H9N2 viruses. The sequence identity range is indicated in different colors and above each gene segment. The arrow shows the earliest to the latest isolation date from 2010 to 2013. Colors indicate high (green, 99%–100%), median (yellow, 96%–98%) and low relative similarities (red, 94%–95%) for each indicated gene after the sequence identity was normalized in the matrix to have a mean of 0 and variance of 1.
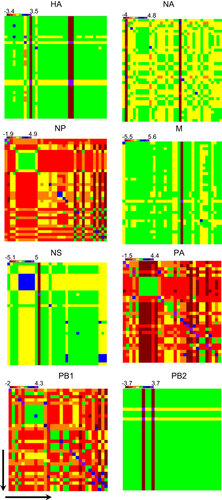
Figure 4 Unrooted maximum-likelihood phylogenetic trees for the (A) HA, (B) NA, (C) M, (D) NP, (E) NS, (F) PA, (G) PB1 and (H) PB2 genes of all analyzed H9N2 viruses. The trees were generated by minimum evolution analysis with maximum likelihood, using the Tamura-Nei model in MEGA (version 5.1). Full-length sequences with complete open reading frames were used for the phylogenetic analyses. Bootstrap values from 1000 replicates are indicated below the branches, and bootstrap values greater than 60 are shown. The scale bar represents the distance units between sequence pairs. Representative prototypes viruses from different H9N2 lineages are indicated in red, the human H9N2 isolate from Bangladesh is indicated in fuchsia and the H7N3 isolate from Pakistan is indicated in blue. The possible H5N1 recombinant isolates in the PB1 gene are indicated in green.
Figure 5 (A) Map showing the hypothesized and proposed routes taken by avian influenza H9N2 virus from Hong Kong to Bangladesh, based on H9N2 isolated from specific locations and years. Numbers 1–6 represent the sequence of events that took place after H9N2 was identified in Hong Kong in 1997.
H9N2 spread from Hong Kong to the United Arab Emirates (UAE; Middle East) and Pakistan and from Pakistan to Japan during and after 1997 via pet trade.
After establishing a separate lineage, H9N2 spread from the UAE to Pakistan during early 2000.
The Iranian H9N2 lineage moved to Pakistan via the cross-border poultry trade.
During 2004, H7N3/H9N2 reassortment occurred in Pakistan, followed by H9N2/H7N3 reassortment involving H9N2 lineages from the UAE and Iran.
H9N2/H7N3 reassortants spread to terrestrial poultry in India via the cross-border trade.
H9N2/H7N3 reassortants spread from India to Bangladesh, became firmly established and evolved into a distinct sublineage. (B) Evolution of Bangladeshi H9N2 through inter- and intrasubtypic reassortment between different H9N2 lineages and HPAI H7N3 and HP H5N1. H9N2 and H7N3 viruses are represented by black and red ovals, with each containing horizontal bars that represent the eight viral gene segments (starting from the top: PB2, PB1, PA, HA, NP, NA, M, NS). To illustrate the gene ancestry through reassortment events, the gene segments in descendent H9N2 viruses are colored according to their corresponding source viruses. Mideast B and C lineages of H9N2 were defined by lineage assignment by extended learning (LABEL).Citation17 In 1997, the prototypical H9N2 belonging to the G1 clade was identified in Hong Kong, and this lineage continued to circulate in poultry species until 2000. During 2000–2003, in the UAE and Iran, H9N2 formed distinct lineages with the G1 background that were identified by phylogenetic analyses. In 2004, reassortment occurred between H9N2/G1 and H7N3, thereby generating several true and hypothetical reassortants (broken oval). In 2004, H9N2/G1 also reassorted with H5N1 (green) to generate an H9N2 reassortant with the NS gene derived from H5N1. From 2009, many inter- and intrasubtypic reassortants between the H9N2 lineages from Dubai and Iran with H7N3 from Pakistan emerged to form several H9N2 influenza virus genotypes that moved through India to Bangladesh.

Homology and phylogenetic analyses
To study the genetic diversity and origin of H9N2 viruses circulating in various terrestrial poultry in LBMs and farms in Bangladesh, we sequenced the full genomes of 37 different H9N2 viruses isolated from different LBMs in Bangladesh from August 2010 to December 2013 ( and Supplementary Table S2). Using a combination of sequence identity matrices generated through MegAlign and BLAST, the full genome of each virus isolate was used for the homology analysis. All recently sequenced H9N2 viruses had very high nucleotide homology (>99%) to those sequenced from the previous years in Bangladesh (), which suggests that H9N2 viruses are evolving as a very homogenous population. Furthermore, all of the analyzed gene segments had >97% nucleotide sequence homology with the human H9N2 isolate A/Bangladesh/0994/2011 (), revealing a genetic conservation between the human and avian H9N2 viruses. Comparison of the Bangladeshi H9N2 viruses to other non-Bangladeshi H9N2 viruses revealed that all of the H9N2 gene segments had >96% nucleotide homology to H9N2 viruses circulating in India during the late 2000s, which suggested a possible transmission pathway (). Nucleotide sequence comparison of the Bangladeshi H9N2 viruses with prototype viruses from other lineages from China and the Middle East revealed that the HA gene had 93% homology to Iran-B102 (Mideast B clade, the neuraminidase (NA) and polymerase basic 2 (PB2) genes had 93% and 94% homology, respectively, to A/Quail/Dubai/303/2000 (H9N2) (Mideast C clade)). Notably, the internal genes (Matrix (M), NS, nucleoprotein (NP), polymerase acidic (PA) and polymerase basic 1 (PB1)) had >96% sequence homology to HPAI (H7N3) A/Chicken/Karachi/NARC-100/04 (). Surprisingly, the polymerase gene PB1 shared 97.8% sequence homology with the recent HPAI H5N1 isolate A/Chicken/Bangladesh/12VIR-7140-7/2012. The high sequence homology between the PB1 genes of the H9N2 and H5N1 viruses suggests reassortment between these subtypes in the recent past.
Table 1 H9N2 influenza viruses isolated from live-bird markets in Bangladesh from August 2010 to December 2013
Table 2 Nucleotide sequence homology of Bangladeshi H9N2 isolates and representative H9N2 viruses
Next, we performed sequence alignment and pairwise comparison of the full gene segments of the 37 H9N2 viruses with respect to each other, and a percent identity matrix was generated for each gene segment. After normalizing the value for a mean of 0 and a variance of 1, we generated a heat map (Figure ). The internal genes NP, PA and PB1 were most divergent, followed by M, NS and NA. The HA and PB2 genes showed very little divergence in all of the analyzed H9N2 viruses, suggesting that they were more conserved than other genes (Figure ).
We constructed phylogenetic trees using the Bangladeshi H9N2 viruses that had been sequenced since 2009 along with representative prototype H9N2 viruses from the Chinese, Middle Eastern and Eurasian lineagesCitation17 and with H5N1 and H7N3 viruses that were identified to be very similar to Bangladeshi H9N2 viruses. The overall topology of all trees that were generated for each gene segment showed that the Bangladeshi H9N2 viruses clustered with the H9N2 viruses isolated from India, Pakistan and the Middle East (Figure ). In the HA tree, all of the H9N2 viruses formed a well-supported monophyletic cluster (99% bootstrap value) in the Mideast B sublineage (Figure ) and shared a nucleotide sequence homology of >93% with A/Chicken/Iran/B102/2005, the reference strain of the G1_Mideast B sublineage (). Phylogenetic analyses of the NA genes revealed that they had close nucleotide similarity (92.2%) () with A/Quail/Dubai/303/2000 (H9N2), the prototype virus for the G1_Mideast C_clade and formed a well-supported monophyletic cluster (99% bootstrap value) with it (Figure ). The close clustering of all the viruses with the A/Chicken/Iran/B102/2005 and A/Quail/Dubai/303/2000 implies that the surface glycoprotein might originate from two different H9N2 sublineages. Additionally, both the HA and the NA genes had ∼98% nucleotide homology with the human H9N2 isolate (A/Bangladesh/0994/2011) (), which suggests a common origin for human and avian H9N2 viruses. This theory is supported by the clustering of genes from human H9N2 viruses with the avian H9N2 viruses in the phylogenetic tree, which has a bootstrap value of >99% ().
The phylogenetic trees of the M, NP, and NS genes also clustered and were closely related to A/Chicken/Karachi/NARC-100/2004, an H7N3 isolate from Pakistan (). The H9N2 NS, NP, and M genes had nucleotide sequence similarity of ∼96% with the H7N3 isolate (). As observed for the HA and NA genes, the NS, NP and M genes from the human H9N2 virus A/Bangladesh/0994/2011 clustered with the sequenced avian H9N2 viruses () with a nucleotide sequence similarity of 97% for the NS and NP genes and of 99% for the M gene ().
Phylogenetic analysis of the polymerase genes showed that the topologies of the PA, PB1 and PB2 genes were very similar and that they clustered with H9N2 viruses isolated from India, Pakistan, and the Middle East (). The PA and PB1 genes from all H9N2 viruses sequenced from 2009 clustered with/Chicken/Karachi/NARC-100/2004 (H7N3) and shared nucleotide homology of >95% with the H7N3 isolate (). Interestingly, for the PB1 gene, a single H5N1 isolate, A/Chicken/Bangladesh/12VIR-7140-16/12, grouped with the H9N2 viruses isolated during 2012–2013 and had ∼98% sequence homology with the H9N2 viruses (). This finding suggests the occurrence of a reassortment event that occurred between low-pathogenic avian influenza H9N2 and HPAI H5N1 within the last 2 years. The PB2 gene for all H9N2 viruses clustered with the H9N2 viruses isolated from India and the Middle East and grouped with A/Quail/Dubai/303/2000 (H9N2), the prototypical H9N2 virus for the G1_Mideast C clade.
Our phylogenetic analyses and the similarities in nucleotide sequences suggest that the genomes of the H9N2 viruses currently circulating in Bangladesh poultry are derived from at least three different lineages and two different subtypes (H9N2/G1_Mideast B/C and H7N3). The HA gene may have originated from the G1_Mideast B clade, the NA and PB2 genes may have originated from the G1_Mideast C clade, and the internal genes NP, NS, M, PA and PB1 may have originated from a prior reassortment event with HPAI H7N3 in Pakistan ().
Molecular analysis
To determine whether the Bangladeshi H9N2 viruses had evolved and acquired newer genetic markers associated with mammalian pathogenicity, virulence and adaptation to new hosts since our previous study on Bangladeshi H9N2 viruses isolated from 2009 to 2011,Citation4 we compared all genes of the Bangladeshi H9N2 viruses sequenced in 2009–2011 with those sequenced in 2012–2013 (Supplementary Table S3). As reported in our previous study, most of the mammalian adaptation and virulence markers have become fixed (Supplementary Table S2), except for the following markers. Some of the PB1 markers observed in 2009–2011Citation4 were notably missing in the present H9N2 viruses (Supplementary Table S3), indicating a selection process. Interestingly, only 2 of the 37 2012-13 Bangladesh H9N2 isolates had substitutions in the PA gene compared to the 2009–2011 Bangladesh H9N2 viruses: A/Chicken/Bangladesh/19145/13 with the D55N substitution and A/Environment/Bangladesh/20199/13 with the S65Y substitution, which are both mammalian adaptation markers. The second polypeptide encoded from the PB1 gene, PB1-F2, was full length (90 amino acids). Notably, the N66S substitution, which is thought to increase virulence in mice, was identified in 15 of the 31 analyzed H9N2 isolates (Supplementary Table S3). This substitution was identified in only one isolate in the previous study, which suggests that H9N2 viruses are evolving rapidly to adapt to mammalian species. None of the Bangladesh H9N2 isolates that were analyzed in the current study had the prototypical E627K mammalian adaptation in the PB2 gene.
DISCUSSION
H9N2 viruses rapidly evolve and acquire mutations that can facilitate their ability to replicate in mammalian hosts. Some recent H9N2 viruses can replicate in mice and ferrets without prior adaptations,Citation25,Citation26,Citation27,Citation28 and some H9N2 isolates have already crossed to pigs and humans.Citation27,Citation29 Additionally, in recent years, these H9N2 viruses have enabled other subtypes to evolve and infect humans by donating their internal genes.Citation8,Citation9,Citation10 These observations highlight the importance of the H9N2 subtype and in studying how it evolves and contributes to the evolution and pathogenesis of other influenza subtypes.
In Bangladesh, AIV subtypes H9N2 and H5N1 have been regularly isolated from the domesticated poultry sold in LBMs since 2009. In the current study period (2010–2013), we identified H9N2 and H5N1 as the two dominant subtypes isolated from asymptomatic poultry species (chickens and quails) traded in LBMs. Other subtypes, such as HP H7N3 from neighboring Pakistan and LP-H7N9 from China, were not isolated. Molecular characterization of the 37 representative H9N2 viruses showed that they possess multiple genomic markers that facilitate host switching from birds to humans and affect the pathogenesis and virulence capabilities of these viruses (Supplementary Table S3). Heat maps generated from similarity scores calculated during nucleotide alignment showed that the NP, PA and PB1 genes are more diverse: the M, NS and NA genes are evolving at a slower rate; and the evolution of HA and PB2 genes has stabilized in recent years.
Phylogenetic analyses revealed that the H9N2 currently circulating in Bangladesh is mostly homogenous and possesses Pakistani-origin, H7N3-like NS, NP, MP, PA, and PB1 genes, an A/Chicken/Iran/B102/2005 (H9N2)-like HA gene, and A/Quail/Dubai/303/2000 (H9N2)-like NA and PB2 genes. This genotype strongly suggests an intrasubtype reassortment that took place in the early 2000s, when these isolates were first detected in the Middle East and Pakistan. The current H9N2 genotype replaced the previous H9N2 genotype (2009–2011), whose gene constellation was similar to that of the current genotype, but possessed only the NS, PA and PB1 genes from HP H7N3. We also found that 2 H9N2 genotypes with internal genes from H7N3 previously circulated in Bangladeshi poultry, but were quickly replaced, which suggests that those genotypes were not fit and not dominant (Figure ).
Our report suggests that the current Bangladeshi H9N2 viruses have emerged from multiple reassortment events that occurred over time between multiple H9N2 sublineages originating from the prototypical G1 lineage and from HP H7N3 isolated from Pakistan. The continued evolution of these H9N2 viruses, with the steady accumulation of mutations in their genomes, has led to the increasing differentiation of Bangladeshi H9N2 viruses from other H9N2 lineages circulating in neighboring countries, leading to the generation of a new H9N2 sublineage in chicken and quail hosts in Bangladesh.
Although the 37 H9N2 viruses were collected in different markets over a 2-year period, the full genomes of all of the viruses clustered together phylogenetically (Figure ), suggesting that they originated from the same source and are uniformly evolving. However, comparison of individual gene segments, as observed in the heat map (Figure ), shows that each gene segment of the H9N2 genome is evolving at a different rate. It is likely that Bangladeshi H9N2 viruses are evolving to adapt and replicate efficiently in the poultry host because genes mostly comprising the nucleoprotein complex (NP, PA and PB1) are the most heterogeneous. Residues in the genes forming the nucleoprotein complex play a vital role in host range, virulence, and replication in the mammalian host;Citation19,Citation30,Citation31 therefore, heterogeneity may be part of the evolutionary process for mammalian adaptation. This premise is consistent with our molecular analysis, which revealed that the NP, PA and PB1 genes have begun to acquire several mammalian host-specific markers (Supplementary Table S3).
Our phylogenetic analysis reveals H7N3 and G1-lineage H9N2 from Pakistan and the Middle East, respectively, as possible sources and origins of the Bangladeshi H9N2 viruses. The real conundrums are how G1-lineage H9N2 AIV was transmitted from Hong Kong to Pakistan or the Middle East because there is no known migratory flyway between China and the Middle East via PakistanCitation21 and how the H9N2/H7N3 reassortant was transmitted from Pakistan to Bangladesh. Our combination of phylogenetic analyses, molecular characterization and speculation identifies the possible evolutionary pathway for current Bangladesh H9N2 viruses (Figure ).
Avian influenza surveillance shows that G1-like viruses may have been introduced into Pakistan and the Middle East after 1997Citation32 (Figure ) and could have continued to circulate in the poultry host for a few years, eventually leading to a well-defined genetic subgroup (H9N2_Mideast A/B/C/D lineages), as reported previously.Citation16,Citation17 Multiple reassortment events between lineages (A, B, C and D) and the mixing of HPAI H5N1 and/or H7N3 in Pakistan led to further reassortment events, generating viruses with varying genotypes (Figure ). This notion is consistent with the sequence information from H9N2 viruses isolated from 2005 to 2008 in Pakistan.Citation19 None of the viruses clustered into a single H9N2 sublineage, but they showed evidence of reassortment with HPAI H5N1 (e.g., the A/Chicken/Pakistan/UDL-1/05 H9N2/H5N1 reassortant) and HPAI H7N3 (e.g., the A/Chicken/Pakistan/UDL-1/06-H9N2/H7N3 reassortant).Citation19 The poultry trade and possibly the pet trade may have played an important role in the introduction of H9N2 from Hong Kong to the Middle East and Pakistan because the Arabian Peninsula is one of the most important import markets in the world.Citation16,Citation33
Phylogeny-based geographic association analysis of a single Bangladesh H9N2 virus isolated in 2006 (A/Chicken/Bangladesh/VP01/06-H9N2) showed that the internal genes of this virus were from H7N3 isolated in Pakistan.Citation5 This finding is consistent with the available evidence to date, which suggests that mixing of H7N3 and H9N2 viruses has only been detected in Pakistan.Citation19 We can speculate that the H9N2 viruses were introduced into backyard poultry in Bangladesh by cross-border trading or smuggling from Pakistan, presumably via India, during the early 2000s, because ∼18% of backyard chickens tested during 2000–2003 were seropositive for AIVs.Citation1,Citation34 Therefore, Pakistani-origin H9N2 viruses with various combinations of H7N3 internal gene constellations are likely being continually introduced into Bangladesh poultry via India (Figure ). This notion is supported by the fact that the 2006 Bangladeshi H9N2 virus had G1-lineage surface glycoproteins (HA and NA) and all of the internal genes from H7N3 (Figure ), but the 2009–2011 Bangladeshi H9N2 viruses had the NS, PA and PB1 genes from H7N3, which were eventually replaced by the current circulating genotype.
During our earlier avian influenza surveillance, we found increased cocirculation of H9N2 and HP H5N1 in the same host without reassortment. However, during this study period, we observed that a PB1 H5N1–H9N2 reassortant emerges. Molecular assessment of the Bangladeshi H9N2 viruses during this study period and during the previous studyCitation4 show that these H9N2 viruses have acquired molecular markers throughout their genomes that facilitate replication and transmission in the mammalian host (Supplementary Table S3). This finding is similar to what has been observed in other neighboring countries in the recent past, but why this situation is presently arising in Bangladesh and what are the conditions that facilitate the evolution of H9N2 needs to be critically monitored.
In Bangladesh, most poultry raised in backyards and small-scale commercial poultry farms are sold in LBMs. In our ongoing avian influenza surveillance, H9N2 with H7N3 internal genes and HP H5N1 were the only subtypes isolated from domesticated poultry traded in these LBMs, and these subtypes were not found in wild birds, which suggests that LBMs are a breeding ground for avian influenza. This finding is consistent with previous findings showing that the trade of chickens and quails in LBMs has played a pivotal role in the emergence of a novel influenza virus.Citation7,Citation9,Citation35 Recent studies also show that H9N2 viruses might be ‘incubators’ or ‘enablers’ for influenza viruses with a wild-bird origin (H7N9, H10N8) that infect humansCitation7,Citation9,Citation10,Citation36 and that any reassortment between poultry H9N2 viruses and HP viruses could trigger increased ability to infect humans. These factors, combined with the tropical climate in this region, facilitate long-term survival, proliferation and transmission among different poultry species and from poultry to humans. These factors also provide ample opportunities for new and prevailing influenza viruses such as H9N2 and H5N1 to recombine and generate novel viruses with varying host specificity. Gene segments from H9N2, HP H7N3 and HP H5N1 continue to circulate in LBMs in Bangladesh, serving as an optimal mixing pot for the generation of novel influenza subtypes and thereby making Bangladesh a hotspot for avian influenza outbreaks. Therefore, the preventive measures that were effective in the 1997 Hong Kong H5N1 outbreaks should be used in LBMs in Bangladesh to reduce the possibility of an influenza outbreak. The changes that greatly reduced the prevalence of avian influenza in live bird markets in Hong Kong included the ruling of no carryover of birds from one day to the next and daily stall cleaning. Birds remaining at the end of the day are killed, ‘dressed’, chilled and sold mainly to restaurants at moderately reduced prices. In the absence of such interventions, the generation of novel H9N2 clades with varying host range and pathogenicity is unavoidable and can potentially become a major public health concern. Thus, there is a need for continual monitoring and surveillance of H9N2 viruses in Bangladesh to identify the evolution and incremental adaptation of viruses to humans to be better prepared for potential future outbreaks.
Supplementary Figure 1
Download MS Word (47.5 KB)We thank Jerry Parker, Richard Elia and Mark Weilnau for maintaining the influenza database at St Jude Children’s Research Hospital, Memphis, USA; James Knowles for administrative assistance; and Vani Shanker for scientific editing. This work was supported by Contract NO HHSN266200700005C from the National Institute of Allergy and Infectious Disease, National Institutes of Health, Department of Health and Human Services, and by the American Lebanese Syrian Associated Charities. The nucleotide sequences obtained in this study have been deposited in GenBank and are available for download using accession numbers KJ643594–KJ643868.
Supplementary Information for this article can be found on Emerging Microbes & Infections's website (http://www.nature.com/emi/).
- Biswas PK, Christensen JP, Ahmed SS et al.Avian influenza outbreaks in chickens, Bangladesh. Emerg Infect Dis2008;14: 1909–1912.
- Monne I, Yamage M, Dauphin G et al.Reassortant avian influenza A(H5N1) viruses with H9N2-PB1 gene in poultry, Bangladesh. Emerg Infect Dis2013;19: 1630–1634.
- Negovetich NJ, Feeroz MM, Jones-Engel L et al.Live bird markets of Bangladesh: H9N2 viruses and the near absence of highly pathogenic H5N1 influenza. PLoS ONE2011;6: e19311.
- Shanmuganatham K, Feeroz MM, Jones-Engel L et al.Antigenic and molecular characterization of avian influenza A(H9N2) viruses, Bangladesh. Emerg Infect Dis2013;19.doi: https://doi.org/10.3201/eid1909.130336 .
- Parvin R, Heenemann K, Halami M, Chowdhury E, Islam MR, Vahlenkamp T.Full-genome analysis of avian influenza virus H9N2 from Bangladesh reveals internal gene reassortments with two distinct highly pathogenic avian influenza viruses. Arch Virol2014;159: 1651–1661.
- Chakraborty A, Arifeen SE, Streafield PK.Outbreak of mild respiratory disease caused by H5N1 and H9N2 infections among young children in Dhaka, Bangladesh. Health Sci Bull2011;9: 5–12
- Chen H, Yuan H, Gao R et al.Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: a descriptive study. Lancet2014;383: 714–721.
- Gao R, Cao B, Hu Y et al.Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med2013;368: 1888–1897.
- García-Sastre A, Schmolke M.Avian influenza A H10N8? A virus on the verge? Lancet2014;383: 676–677.
- Liu D, Shi W, Gao GF.Poultry carrying H9N2 act as incubators for novel human avian influenza viruses. Lancet2014;383: 869.
- Butt AM, Siddique S, Idrees M, Tong Y.Avian influenza A (H9N2): computational molecular analysis and phylogenetic characterization of viral surface proteins isolated between 1997 and 2009 from the human population. Virol J2010;7: 319.
- Ge FF, Zhou JP, Liu J et al.Genetic evolution of H9 subtype influenza viruses from live poultry markets in Shanghai, China. J Clin Microbiol2009;47: 3294–3300.
- Guan Y, Shortridge KF, Krauss S et al.H9N2 influenza viruses possessing H5N1-like internal genomes continue to circulate in poultry in southeastern China. J Virol2000;74: 9372–9380.
- Guan Y, Shortridge KF, Krauss S, Webster RG.Molecular characterization of H9N2 influenza viruses: were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proc Natl Acad Sci USA1999;96: 9363–9367.
- Xu KM, Li KS, Smith GJ et al.Evolution and molecular epidemiology of H9N2 influenza A viruses from quail in southern China, 2000 to 2005. J Virol2007;81: 2635–2645.
- Fusaro A, Monne I, Salviato A et al.Phylogeography and evolutionary history of reassortant H9N2 viruses with potential human health implications. J Virol2011;85: 8413–8421.
- Shepard SS, Davis CT, Bahl J, Rivailler P, York IA, Donis RO.LABEL: fast and accurate lineage assignment with assessment of H5N1 and H9N2 influenza A hemagglutinins. PLoS ONE2014;9: e86921.
- Toroghi R, Momayez R.Biological and molecular characterization of Avian influenza virus (H9N2) isolates from Iran. Acta Virol2006;50: 163–168.
- Iqbal M, Yaqub T, Reddy K, McCauley JW.Novel genotypes of H9N2 influenza A viruses isolated from poultry in Pakistan containing ns genes similar to highly pathogenic H7N3 and H5N1 viruses. PLoS ONE2009;4: e5788.
- Tosh C, Nagarajan S, Behera P et al.Genetic analysis of H9N2 avian influenza viruses isolated from India. Arch Virol2008;153: 1433–1439.
- Wetlands International. What are Flyways?Fredericton: Wetlands International, 2014.Available at http://wpe.wetlands.org/Iwhatfly (Accessed 10 October 2014).
- Marinova-Petkova A, Feeroz MM, Rabiul Alam SM et al.Multiple introductions of highly pathogenic avian influenza H5N1 viruses into Bangladesh. Emerg Microbes Infect2014;3: e11.
- Hall TA.BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser1999;41: 95–98.
- Pavlidis P, Noble WS.Matrix2png: a utility for visualizing matrix data. Bioinformatics2003;19: 295–296.
- Choi YK, Ozaki H, Webby RJ et al.Continuing evolution of H9N2 influenza viruses in Southeastern China. J Virol2004;78: 8609–8614.
- Guo YJ, Krauss S, Senne DA et al.Characterization of the pathogenicity of members of the newly established H9N2 influenza virus lineages in Asia. Virology2000;267: 279–288.
- Li C, Yu K, Tian G et al.Evolution of H9N2 influenza viruses from domestic poultry in Mainland China. Virology2005;340: 70–83.
- Wan H, Sorrell EM, Song H et al.Replication and transmission of H9N2 influenza viruses in ferrets: evaluation of pandemic potential .PLoS ONE2008;3: e2923.
- Xu KM, Smith GJ, Bahl J et al.The genesis and evolution of H9N2 influenza viruses in poultry from southern China, 2000 to 2005 .J Virol2007;81: 10389–10401.
- Chen GW, Chang SC, Mok CK et al.Genomic signatures of human versus avian influenza A viruses. Emerg Infect Dis2006;12: 1353–1360.
- Shaw M, Cooper L, Xu X et al.Molecular changes associated with the transmission of avian influenza a H5N1 and H9N2 viruses to humans. J Med Virol2002;66: 107–114.
- Mase M, Imada T, Sanada Y et al.Imported parakeets harbor H9N2 influenza A viruses that are genetically closely related to those transmitted to humans in Hong Kong. J Virol2001;75: 3490–3494.
- Nicita A. Avian influenza and the poultry trade.Washington, DC: The World Bank eLibrary, 2008.Available at http://dx.doi.org/10.1596/1813-9450-4551 (accessed 27 October 2014).
- Alam J KI.Seroprevalence of poultry diseases in native chickens in Bangladesh.In: Proceedings of the 9th Annual Scientific Conference of the Bangladesh Society for Veterinary Education and Research; 6–7 January 2003; Mymensingh, Bangladesh. Bangladesh Society for Veterinary Education and Research: Mymensingh, Bangladesh, 2003, Abstract 11.
- US Defense Threat Reduction Agency. Anticipating the species jump: surveillance for emerging viral threats.Ft Belvoir VA: DTRA, 2010.Available at http://www.hsdl.org/?view&did=716028 (accessed 27 October 2014).
- Yu X, Jin T, Cui Y et al.Coexistence of influenza H7N9 and H9N2 in poultry linked to human H7N9 infection and their genome characteristics. J Virol2014;88: 3423–3431.