
Abstract
Hand, foot and mouth disease (HFMD) is a serious public health problem that has emerged over the past several decades. Pathogen detection by the Chinese national HFMD surveillance system has focused mainly on enterovirus 71 (EV71) and coxsackievirus A16 (CA16). Therefore, epidemiological information regarding the other causative enteroviruses is limited. To identify the pandemic enterovirus in Suzhou, Jiangsu province, China, clinical samples from patients with HFMD were collected from 2012 to 2013 and analyzed. The results revealed that CA16 was the most dominant HFMD pathogen in 2012, whereas CA6 and CA10 were the dominant pathogens in 2013. Phylogenetic analysis revealed that the C4a sub-genogroup of EV71 and the B1a and B1b sub-genogroups of CA16 continued to evolve and circulate in Suzhou. The CA6 strains were assigned to six genotypes (A–F) and the CA10 strains were assigned to seven genotypes (A–G), with clear geographical and temporal distributions. All of the CA6 strains in Suzhou belonged to genogroup F, and there were several lineages circulating in Suzhou. All of the CA10 strains in Suzhou belonged to genogroup G, and they had the same genetic origin. Co-infections of EV71/CA16 and CA6/CA10 were found in the samples, and bootscan analysis of 5′-untranslated regions (UTRs) revealed that some CA16 strains in Suzhou had genetic recombination with EV71. This property might allow CA16 to alter its evolvability and circulating ability. This study underscores the need for surveillance of CA6 and CA10 in the Yangtze River Delta and East China.
Introduction
Hand, foot and mouth disease (HFMD) is a common viral disease that infects infants and children aged <10 years.Citation1 After several large-scale outbreaks of HFMD in the Asia-Pacific region in 1997, HFMD was defined as a notifiable disease in many countries.Citation2 In mainland China, large-scale outbreaks of HFMD occurred in Linyi in 2007Citation3 and in Fuyang in 2008.Citation4 Therefore, HFMD has drawn considerable attention from the Ministry of Health of China. HFMD was classified as a category C notifiable infectious disease, and a virological surveillance system was set up in 2008.Citation4,Citation5 Because the system mainly detects enterovirus 71 (EV71) and coxsackievirus A16 (CA16), information on the geographical distribution and epidemiological profiles of other enterovirus types in China is relatively limited.
Human enterovirus group A (HEV-A) members, which belong to the Picornaviridae family, are the major etiologic agents of HFMD. The HEV-A group includes CA2, CA3, CA4, CA5, CA6, CA7, CA8, CA10, CA12, CA14, CA16 and EV71, and among them, EV71 and CA16 are the typical causative pathogens of HFMD.Citation6 Children suffering from EV71 infection might develop severe complications such as acute flaccid paralysis, myocarditis or even fatal encephalitis. However, compared with EV71, CA16-associated HFMD is usually mild and benign.Citation7,Citation8,Citation9,Citation10 CA16- or EV71-infected adults rarely present clinical manifestations. Recently, outbreaks of other HEV-A strains have been reported frequently, such as outbreaks of CA6 and CA10 in Singapore, Finland, Taiwan district and Japan.Citation11,Citation12,Citation13,Citation14 In mainland China, the prevalence of CA6, CA10 and other enterovirus strains has grown to epidemic proportions in some areas.Citation15,Citation16,Citation17,Citation18,Citation19,Citation20 Clinical manifestations of CA6 include mild herpangina or HFMD disease, characterized by vesicles and ulcers around the uvula, papules and vesicles on the dorsal aspects of the hands and feet, or acute fever.Citation11,Citation19,Citation20 CA6 can also infect adults with severe clinical presentations.Citation20,Citation21 Therefore, it is important to study the epidemiology of CA6 in detail.
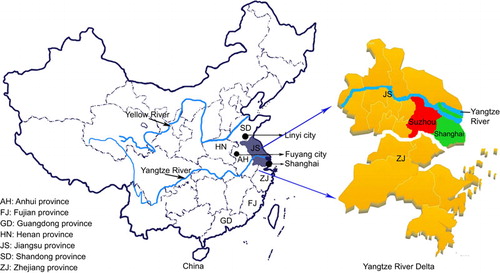
In this study, we analyzed the epidemiology of HFMD in Suzhou, located in the Jiangsu province. The Jiangsu province shares a border with the Anhui province in the west, and with the Shandong province in the north. Large-scale HFMD outbreaks occurred in the Shandong province in 2007 and in the Anhui province in 2008. Therefore, the Jiangsu province is located at a vital geographical position for the transmission and spread of HEV-As to other regions of the Yangtze River Delta, such as the Shanghai and Zhejiang province (). The city of Suzhou, which is an important economic and transportation center in the Yangtze River Delta, might function as a wind vane in the epidemiology of HEV-As in the Yangtze River Delta. Investigating HFMD epidemics in Suzhou might aid the prevention of HFMD not only in the Jiangsu province, but also in the Yangtze River Delta, or even in East China.
MATERIALS AND METHODS
Clinical information and specimen collection
Sentinel surveillance was performed from 2012 to 2013, and the sentinel pediatricians were requested to collect clinical samples from patients presenting HFMD manifestations. Diagnosis was performed according to the diagnostic criteria defined by the Ministry of Health of China. Generally, children who had fever or one of the following features: maculopapular or vesicular rash on the palms and/or soles, or vesicles and ulcers around the uvula, were diagnosed as HFMD-positive. Severe manifestations of the disease included encephalitis, meningitis, acute flaccid paralysis or even death. Medical records of each patient, including demographic data, clinical symptoms and clinical diagnosis, were obtained. Of the throat or rectal swabs collected by the Suzhou Industrial Park Centers for Disease Control and Prevention (SZIPCDC) from 2012 to 2013, 109 samples were chosen randomly for further analysis. Written informed consents were obtained from each patient or their guardians, and the study was approved by the Human Ethics Committee of the SZIPCDC.
Table 1 PCR and sequencing primers used in this study
Table 2 Demographics of all participants (n=109)
Sample processing, RNA extraction and reverse transcription polymerase chain reaction (RT-PCR)
The clinical samples were thawed and frozen for three times. Viral RNA was then extracted using the QIAamp Viral RNA Kit (Qiagen, Hilden, North Rhine-Westphalia, Germany). RT-PCR was performed using the ABI RT-PCR Kit (Life techonologies, Carlsbad, California, USA). All procedures were performed by following the instructions strictly. The cDNA was prepared for the subsequent experiments.
Detection and genotyping of the HEV-As
To identify and classify the clinical samples, a semi-nested PCR, targeting the 5′-untranslated region (UTR) sequences of different enterovirus types was performed as described previously.Citation22 The PCR product was harvested for gel electrophoresis and purification. The amplicons were sequenced, and the resulting sequence was analyzed using the Basic Local Alignment Search Tool.
For the HEV-A-positive samples, genotyping based on semi-nested PCR, targeting the partial VP1 (gene for encoding viral capsid protein 1) sequence, was performed. The primer pairs, 222/224 and AN88/AN89 were applied,Citation23 and the products were harvested for gel electrophoresis and purification. The primers' sequences are listed in .
Sequencing and phylogenetic analysis of different enterovirus types
The amplicons were sequenced using an ABI 3730xl Genetic Analyzer (Life techonologies, Carlsbad, California, USA) and the Sanger dideoxy sequencing method. The sequences of VP1s and 5′-UTRs have been submitted to GenBank under the following accession numbers, from KP164031 to KP164252.
Multiple sequence alignments were performed for the partial VP1 sequences of the Suzhou samples and for sequences retrieved from the GenBank database, using the ClustalW program.
Phylogenetic trees were constructed using the neighbor-joining statistical method with the Kimura two-parameter nucleotide substitution model in 1000 replicates. Calculations of pairwise nucleotide identities were performed by including all three codon positions and completely deleting all alignment gaps. All calculations were performed using the MEGA 5.2 software.
Recombination analysis
Alignment of the all the 5′-UTR sequences was performed using the ClustalW package in MEGA 5.2. Potential recombinations between different HEV-A strains were scanned using the SimPlot software package (version 3.5.1, Johns Hopkins University, Baltimore, Maryland, USA). Bootscan analysis was performed in each 60-nucleotide window using the Kimura two-parameter substitution model with a transition/transversion ratio of 2. The window was advanced successively along the 5′-UTR alignment in 10-nucleotide increments.
RESULTS
Study participants and epidemiologic characteristics
In this study, we enrolled 109 infants and children from the city of Suzhou, Jiangsu province (). The age of the participants ranged from five months to nine years old, and the study population included 72 males and 37 females. The participants were divided into six age groups. Children aged between one and four years accounted for a large percentage (83/109, 76.2%) of the study population.
From 2012 to 2013, 109 samples were identified (30 cases in 2012 and 79 cases in 2013) in Suzhou. In 2012, CA16 infections accounted for most of the cases (83.3%; 25/30) while only three cases of EV71 infection and one case of CA6 infection were identified. No CA10 infection was identified in 2012. In 2013, the pathogen prevalence spectrum changed. The CA10 (22 cases in 2013) and CA6 strains (32 cases in 2013) accounted for most of the cases (3.3% of all cases in 2012; 68.4% of all cases in 2013). However, only 18 cases of CA16 infection and six cases of EV71 infection were detected in 2013. Here the results showed the percentage of CA16 and EV71 dropped down from 2012 to 2013, which accorded with the annual survey results from SZIPCDC: in 2012, EV71 and CA16 covered 78% of the enterovirus positive samples in Suzhou, but in 2013, EV71 and CA16 covered only 30% of the enterovirus positive samples (data not published).
Co-infections of different HFMD viruses were also detected in the clinical samples. In 2012, one case involved both EV71 and CA16, and in 2013, one case involved both CA6 and CA10.
EV71 and CA16 shared the same seasonality, as did CA6 and CA10. The number of CA16 cases in 2012 or 2013 reached a peak during the warm seasons (from April to August), while CA10 or CA6 infections were most prevalent in June, indicating that optimal temperatures are vital for the transmission of HFMD.
Sustained transmission of EV71 (sub-genogroup C4a) and CA16 (sub-genogroups B1a and B1b) in Suzhou
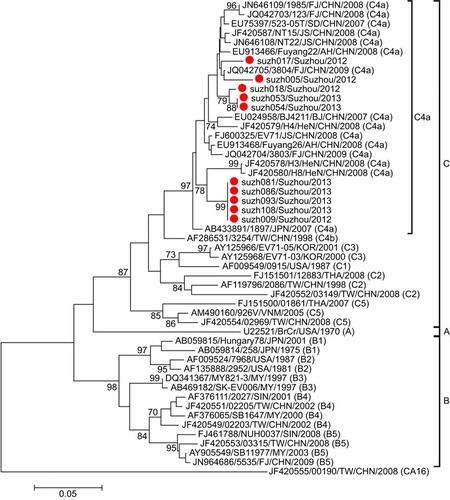
Phylogenetic analysis was performed for the partial VP1 sequences of EV71 and CA16 from both Suzhou isolates and other isolates. The results revealed that the two viruses circulated continuously in 2012 and 2013. Genotyping revealed that the pattern of endemic circulation was as follows: sub-genogroup C4a for EV71 and sub-genogroups B1a and B1b for CA16. The C4a evolutionary branch of EV71 () was responsible for most of the EV71 cases reported in Suzhou. The CA16 isolates from Suzhou belonged to the B1a and B1b evolutionary branches (). Together, C4a of EV71, B1a and B1b of CA16 are the predominant genotypes circulating in mainland China.Citation6
Several lineages of CA6 circulated in Suzhou and shared high sequence similarity with the Shanghai isolates
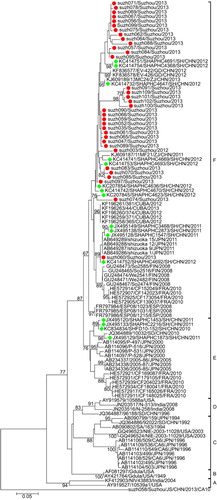
Multiple nucleotide sequence alignments were performed for the partial VP1 sequences from Suzhou isolates and other isolates retrieved from the GenBank database. Phylogenetic analysis revealed that the CA6 strains could be assigned to six genogroups (A–F), with specific temporal and spatial distributions (). Basically, the tree was constructed as described in a previous study.Citation24 All the isolates in the current study were assigned to genogroup F. Genogroup F comprised strains from China isolated in 2011 and 2012, and some latest isolated foreign strains. Most of the Shanghai strains belonged to genogroup F, and the rest were assigned to genogroup E. Genogroups A–D mainly comprised some earlier isolated strains from all over the world. Genogroup E mainly comprised some Shanghai strains and some Japanese and French strains.
The Suzhou isolates formed several independent clusters in the phylogenetic tree, suggesting that several different transmission chains of CA6 circulated in Suzhou. Within genogroup F, the Suzhou strains could be further classified into several sublineages. The pairwise distance among all the Suzhou CA6 strains ranged in divergence from 0.000 to 0.141. The Shanghai strains (obtained from GenBank) showed high sequence similarities with the Suzhou isolates (), and most of the Shanghai isolates branched together with the Suzhou isolates in the phylogenetic tree. This suggested that transmission of CA6 had occurred along the Yangtze River Delta, and this might even facilitate the transmission of CA6 in East China.
CA10 strains in Suzhou had the same genetic origin as most of the Chinese CA10 isolates
The CA10 strains could be assigned to seven genogroups (A–G), with specific temporal and spatial distributions (). Basically, the phylogenetic tree was constructed as previously described.Citation16 Genogroup F mainly included earlier isolated Chinese strains while genogroup G included the latest isolates from mainland China. Genogroups A–E mainly included isolates from Russia, France, Japan and America. Some genogroups could be further divided into several sub-genogroups according to their temporal and spatial distributions.
All the CA10 strains isolated in this study were assigned to genogroup G (), and together, they formed a cluster with a bootstrap value of 86%, suggesting that the CA10 strains circulating in Suzhou were quite similar to each other in sequence and that they might have the ‘founder effect’. Most of the latest Chinese CA10 isolates (including the Suzhou CA10 isolates in this study) clustered together as genogroup G with a high bootstrap value of 99%. The pairwise distance among all the Suzhou CA10 strains ranged in divergence from 0.000 to 0.020, suggesting that the phylogenetic characteristics of the CA10 strains in Suzhou were not as divergent as those of the CA6 strains.
Potential recombination sites were detected in the CA16 isolates
Enteroviruses can undergo extensive genetic recombination. This mechanism is adopted by the virus to generate genetic variation, and thereby, change its profiles. Genomic recombination in enteroviruses was first reported in poliovirus.Citation25,Citation26 Subsequently, many studies have reported that genomic recombination is a relatively frequent event in pandemic enteroviruses and that genetic recombination could occur within different species of enteroviruses.Citation27,Citation28,Citation29,Citation30,Citation31


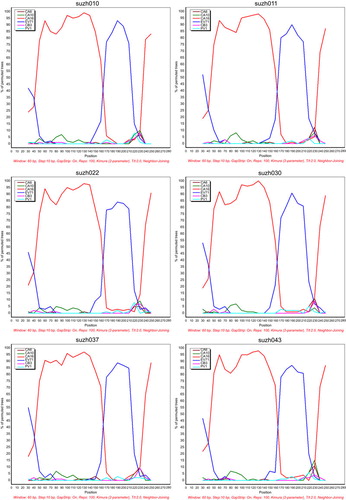
To detect the possible recombination in the Suzhou isolates, 5′-UTR recombination analysis was performed by bootscanning. The following six reference sequences were used in the analysis: EV71, CA6, CA10, CA16, coxsackievirus B3 and poliovirus type 1 (PV1). Of the 44 Suzhou CA16 sequences, 25 showed potential recombinant sites with EV71, of which six strains are shown in . The six isolates, suzh010, suzh011, suzh022, suzh030, suzh037 and suzh043 had breakpoints with EV71 in the 150–180 bp region, implying the potential recombination between the 5′-UTRs of EV71 and CA16. These results are in agreement with previous reports that the 5′-UTRs of enteroviruses are hot spots for recombination, and that the viruses can change their profiles and evolve accordingly.Citation32,Citation33
DISCUSSION
In this study, CA6 and CA10 were identified as the major causative pathogens for HFMD in 2013 in Suzhou. The prevalence of these two strains surpassed that of CA16, which was the most prevalent pathogen for HFMD in 2012 in Suzhou. The two-year surveillance also revealed that CA16 and EV71 were still co-circulating and co-evolving; thus, special attention still should be paid to the prevention of EV71 infections since EV71 infections can be fatal. Also, more HEV-A strains should be included in the surveillance system, and pediatricians should pay more attention to emerging non-CA16 and non-EV71 pathogens for HFMD. Co-infections of EV71-CA16 and C6-CA10 were also detected in the samples collected from Suzhou. Co-infections with different HFMD pathogens have been reported previously,Citation34,Citation35 and such co-infections might lead to the recombination of RNA viruses. In Shanghai, co-infection with EV71 and CA16 was observed in 17.6% of the total CA16 cases between 2009 and 2010;Citation35 such high incidence of co-infection had not been observed in China previously. Because Suzhou is geographically very close to Shanghai, more attention should be paid to the detection of co-infections, as recombination in RNA viruses could change the profiles of the viruses. In accordance with previous findings,Citation36,Citation37 the cases number of boys is larger than that of girls in this study (), and the reasons for that remains to be unclear.
C4a was previously identified as the most prominent EV71 sub-genotype circulating in China.Citation4,Citation5,Citation15 Phylogenetic analysis revealed that the EV71 isolates in this study clustered with other Chinese EV71 strains. Together, they formed a relatively independent cluster (C4a; ), parallel to the cluster of the C4b sub-genogroup. Within the C4a clade, four strains of Suzhou isolates (suzh081, suzh086, suzh093, suzh108) formed a relatively independent cluster with a bootstrap value of 99%, parallel to the branch composed of H3 (JF420587) and H8 (JF420580), differentiating them from the other Suzhou isolates. These results suggested that there were several transmission chains of EV71 in Suzhou.
The CA16 isolates in Suzhou mainly belonged to the B1b sub-genogroup, with two isolates belonging to B1a (). The B1 sub-genogroup is the epidemic strain in mainland China. All the Suzhou CA16 B1b isolates had the same origin on the phylogenetic tree, suggesting the CA16 B1b strains were quite similar to each other in sequence. The percentage of CA16 isolates dropped from 83.3% in 2012 to 22.8% in 2013, suggesting the diminishing epidemics of CA16 in Suzhou.
The CA6 isolates in Suzhou were quite divergent and formed several lineages, suggesting that there are several CA6 transmission chains in Suzhou. Phylogenetic analysis revealed that all the Suzhou isolates clustered with the Shanghai strains, perhaps due to the short geographical distance between the two cities. However, some Suzhou isolates (suzh091, suzh101, suzh100, suzh102, suzh109) formed their own cluster with a high bootstrap value of 98%, indicating that the Suzhou isolates not only shared similarities with the Shanghai isolates, but also evolved to acquire their own genetic characteristics. There were reports of CA6 and CA10 outbreaks in Shanghai between 2010 and 2011.Citation38 Therefore, it is possible that the Shanghai CA6 strains and the Suzhou CA6 strains evolved from the same ancestral strain. Compared to the CA6 isolates, the CA10 isolates in Suzhou were less divergent. All the CA10 isolates in Suzhou belonged to genogroup G, which mainly included isolates from mainland China. The Suzhou CA10 isolates formed one cluster, with a bootstrap value of 86%, suggesting the transmission lines of CA10 is not as variable as CA6 in Suzhou.
Enterovirus could undergo recombination at the non-structural genomic site.Citation25,Citation26,Citation27,Citation28,Citation29,Citation30,Citation31 The 5′-UTR RNA sequence of EV71 contains a cloverleaf structure and some stem-loop structures that are involved in the initiation of positive strand synthesis and genome translation.Citation39,Citation40,Citation41 Previous studies on poliovirus have shown that the 5′-UTR is related to tissue tropism.Citation42 Thus, recombination in the 5′-UTR of the enterovirus might have resulted in the varying virulence toward the host. Since co-infection was found in the samples collected from Suzhou, it is possible that the virus undergoes recombination. Bootscanning analysis revealed that the 5′-UTR sequence of the Suzhou CA16 isolates might have recombination with the EV71 isolates, and such recombination events might change the epidemic trend.
In summary, this study identified the different etiological agents which were responsible for HFMD outbreaks in Suzhou in 2012 and 2013, and the epidemics in Suzhou was also clarified, CA6 and CA10 replaced CA16 as the dominant pathogens for HFMD in 2013. Phylogenetic analysis implied that the CA6, CA10, CA16 and EV71 strains continued to co-circulate and co-evolve in Suzhou. 5′-UTR recombination was observed in the CA16 isolates, and this might have changed the evolvability and genetic characteristics of the enterovirus. Because Suzhou is located in a geographically important position on the Yangtze River Delta, surveillance systems for the other HEV-A strains should be introduced to facilitate the prevention and treatment of HFMD in East China.
This work was supported by grants from the ‘Knowledge Innovation Program’ (NO Y014P31503) and the ‘100 Talent Program’ (NO Y316P11503) from the Chinese Academy of Sciences and the Shanghai Pasteur Foundation. We thank the participating nurses from the Suzhou Industrial Park Centers for Disease Control for their assistance in data and sample collection, and all the participants for taking part in the study.
- Grist NR, Bell EJ, Assaad F.Enteroviruses in human disease. Prog Med Virol1978;24: 114–157.
- Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, Ooi MH.Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis2010;10: 778–790.
- Zhang Y, Tan XJ, Wang HY et al.An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J Clin Virol2009;44: 262–267.
- Zhu Q, Hao Y, Ma J, Yu S, Wang Y.Surveillance of hand, foot, and mouth disease in mainland China (2008–2009). Biomed Environ Sci2011;24: 349–356.
- Yang F, Ren L, Xiong Z et al.Enterovirus 71 outbreak in the People's Republic of China in 2008. J Clin Microbiol2009;47: 2351–2352.
- Zhang Y, Wang D, Yan D et al.Molecular evidence of persistent epidemic and evolution of subgenotype B1 coxsackievirus A16-associated hand, foot, and mouth disease in China. J Clin Microbiol2010;48: 619–622.
- Ho M.Enterovirus 71: the virus, its infections and outbreaks. J Microbiol Immunol Infect2000;33: 205–216.
- Cardosa MJ, Perera D, Brown BA et al.Molecular epidemiology of human enterovirus 71 strains and recent outbreaks in the Asia-Pacific region: comparative analysis of the VP1 and VP4 genes. Emerg Infect Dis2003;9: 461–468.
- Perez-Velez CM, Anderson MS, Robinson CC et al.Outbreak of neurologic enterovirus type 71 disease: a diagnostic challenge. Clin Infect Dis2007;45: 950–957.
- Chang LY, Lin TY, Huang YC et al.Comparison of enterovirus 71 and coxsackie-virus A16 clinical illnesses during the Taiwan enterovirus epidemic, 1998. Pediatr Infect Dis J1999;18: 1092–1096.
- Lo SH, Huang YC, Huang CG et al.Clinical and epidemiologic features of Coxsackievirus A6 infection in children in northern Taiwan between 2004 and 2009. J Microbiol Immunol Infect2011;44: 252–257.
- Wei SH, Huang YP, Liu MC et al.An outbreak of coxsackievirus A6 hand, foot, and mouth disease associated with onychomadesis in Taiwan, 2010. BMC Infect Dis2011;11: 346.
- Fujimoto T, Iizuka S, Enomoto M et al.Hand, foot, and mouth disease caused by coxsackievirus A6, Japan, 2011. Emerg Infect Dis2012;18: 337–339.
- Blomqvist S, Klemola P, Kaijalainen S et al.Co-circulation of coxsackieviruses A6 and A10 in hand, foot and mouth disease outbreak in Finland. J Clin Virol2010;48: 49–54.
- Lu J, Zeng H, Zheng H et al.Hand, foot and mouth disease in Guangdong, China, in 2013: new trends in the continuing epidemic. Clin Microbiol Infect2014;20: O442–O445.
- Tian H, Zhang Y, Sun Q et al.Prevalence of multiple enteroviruses associated with hand, foot, and mouth disease in Shijiazhuang City, Hebei province, China: outbreaks of coxsackieviruses a10 and b3. PLoS One2014;9: e84233.
- Lu QB, Zhang XA, Wo Y et al.Circulation of Coxsackievirus A10 and A6 in hand–foot–mouth disease in China, 2009–2011. PLoS One2012;7: e52073.
- He YQ, Chen L, Xu WB et al.Emergence, circulation, and spatiotemporal phylogenetic analysis of coxsackievirus a6- and coxsackievirus a10-associated hand, foot, and mouth disease infections from 2008 to 2012 in Shenzhen, China. J Clin Microbiol2013;51: 3560–3566.
- Han JF, Xu S, Zhang Y et al.Hand, foot, and mouth disease outbreak caused by coxsackievirus A6, China, 2013. J Infect2014;69: 303–305.
- Downing C, Ramirez-Fort MK, Doan HQ et al.Coxsackievirus A6 associated hand, foot and mouth disease in adults: clinical presentation and review of the literature. J Clin Virol2014;60: 381–386.
- Ben-Chetrit E, Wiener-Well Y, Shulman LM et al.Coxsackievirus A6-related hand foot and mouth disease: skin manifestations in a cluster of adult patients. J Clin Virol2014;59: 201–203.
- Ge S, Yan Q, He S, Zhuang S, Niu J, Xia N.Specific primer amplification of the VP1 region directed by 5′ UTR sequence analysis: enterovirus testing and identification in clinical samples from hand–foot–and–mouth disease patients. J Virol Methods2013;193: 463–469.
- Nix WA, Oberste MS, Pallansch MA.Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J Clin Microbiol2006;44: 2698–2704.
- Puenpa J, Mauleekoonphairoj J, Linsuwanon P et al.Prevalence and characterization of enterovirus infections among pediatric patients with hand foot mouth disease, herpangina and influenza like illness in Thailand, 2012. PLoS One2014;9: e98888.
- Hirst GK.Genetic recombination with Newcastle disease virus, polioviruses, and influenza. Cold Spring Harb Symp Quant Biol1962;27: 303–309.
- Ledinko N.Genetic recombination with poliovirus type 1. Studies of crosses between a normal horse serum-resistant mutant and several guanidine-resistant mutants of the same strain. Virology1963;20: 107–119.
- Zhang Y, Tan X, Cui A et al.Complete genome analysis of the C4 subgenotype strains of enterovirus 71: predominant recombination C4 viruses persistently circulating in China for 14 years. PLoS One2013;8: e56341.
- Oberste MS, Maher K, Pallansch MA.Evidence for frequent recombination within species human enterovirus B based on complete genomic sequences of all thirty-seven serotypes. J Virol2004;78: 855–867.
- Hu YF, Yang F, Du J et al.Complete genome analysis of coxsackievirus A2, A4, A5, and A10 strains isolated from hand, foot, and mouth disease patients in China revealing frequent recombination of human enterovirus A. J Clin Microbiol2011;49: 2426–2434.
- Yip CC, Lau SK, Woo PC, Chan KH, Yuen KY.Complete genome sequence of a coxsackievirus A22 strain in Hong Kong reveals a natural intratypic recombination event. J Virol2011;85: 12098–12099.
- Lukashev AN, Lashkevich VA, Ivanova OE, Koroleva GA, Hinkkanen AE, Ilonen J.Recombination in circulating Human enterovirus B: independent evolution of structural and non-structural genome regions. J Gen Virol2005;86: 3281–3290.
- Lukashev AN, Lashkevich VA, Ivanova OE, Koroleva GA, Hinkkanen AE, Ilonen J.Recombination in circulating enteroviruses. J Virol2003;77: 10423–10431.
- van der Sanden S, van Eek J, Martin DP, van der Avoort H, Vennema H, Koopmans M.Detection of recombination breakpoints in the genomes of human enterovirus 71 strains isolated in the Netherlands in epidemic and non-epidemic years, 1963–2010. Infect Genet Evol2011;11: 886–894.
- Li W, Yi L, Su J et al.Seroepidemiology of human enterovirus71 and coxsackievirusA16 among children in Guangdong province, China. BMC Infect Dis2013;13: 322.
- Yan XF, Gao S, Xia JF, Ye R, Yu H, Long JE.Epidemic characteristics of hand, foot, and mouth disease in Shanghai from 2009 to 2010: enterovirus 71 subgenotype C4 as the primary causative agent and a high incidence of mixed infections with coxsackievirus A16. Scand J Infect Dis2012;44: 297–305.
- Ang LW, Koh BK, Chan KP, Chua LT, James L, Goh KT.Epidemiology and control of hand, foot and mouth disease in Singapore, 2001–2007. Ann Acad Med Singapore2009;38: 106–112.
- Chen SC, Chang HL, Yan TR, Cheng YT, Chen KT.An eight-year study of epidemiologic features of enterovirus 71 infection in Taiwan. Am J Trop Med Hyg2007;77: 188–191.
- Xu M, Su L, Cao L, Zhong H, Dong N, Xu J.Enterovirus genotypes causing hand foot and mouth disease in Shanghai, China: a molecular epidemiological analysis. BMC Infect Dis2013;13: 489.
- Lin JY, Li ML, Shih SR.Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res2009;37: 47–59.
- Thompson SR, Sarnow P.Enterovirus 71 contains a type I IRES element that functions when eukaryotic initiation factor eIF4G is cleaved. Virology2003;315: 259–266.
- Vogt DA, Andino R.An RNA element at the 5′-end of the poliovirus genome functions as a general promoter for RNA synthesis. PLoS Pathog2010;6: e1000936.
- Shiroki K, Ishii T, Aoki T et al.Host range phenotype induced by mutations in the internal ribosomal entry site of poliovirus RNA. J Virol1997;71: 1–8.