
Abstract
The emergence of influenza A virus (IAV) in domestic avian species and associated transmissions to mammals is unpredictable. In the Americas, the H7 IAVs are of particular concern, and there have been four separate outbreaks of highly pathogenic (HP) H7N3 in domestic poultry in North and South America between 2002 and 2012, with occasional spillover into humans. Here, we use long-term IAV surveillance in North American shorebirds at Delaware Bay, USA, from 1985 to 2012 and in ducks in Alberta, Canada, from 1976 to 2012 to determine which hemagglutinin (HA)–neuraminidase (NA) combinations predominated in Anseriformes (ducks) and Charadriiformes (shorebirds) and whether there is concordance between peaks of H7 prevalence and transmission in wild aquatic birds and the emergence of H7 IAVs in poultry and humans. Whole-genome sequencing supported phylogenetic and genomic constellation analyses to determine whether HP IAVs emerge in the context of specific internal gene segment sequences. Phylogenetic analysis of whole-genome sequences of the H7N3 influenza viruses from wild birds and HP H7N3 outbreaks in the Americas indicate that each HP outbreak was an independent emergence event and that the low pathogenic (LP) avian influenza precursors were most likely from dabbling ducks. The different polybasic cleavage sites in the four HP outbreaks support independent origins. At the 95% nucleotide percent identity-level phylogenetic analysis showed that the wild duck HA, PB1, and M sequences clustered with the poultry and human outbreak sequences. The genomic constellation analysis strongly suggests that gene segments/virus flow from wild birds to domestic poultry.
Emerging Microbes and Infections (2015) 4, e35; doi:10.1038/emi.2015.35; published online 17 June 2015
Introduction
While there is general acceptance for the zoonotic origins of influenza A viruses (IAVs) from reservoirs in wild aquatic birds, limited information is available about whether peaks of influenza activity in wild birds correlate with virus spread to other species and about the potential predictive value of surveillance. In the aquatic bird reservoir, the IAVs show a cyclic dominance with peaks of activity for one season followed by low levels of detection.Citation1,Citation2 Long-term surveillance of IAVs in Anseriformes (ducks) in Alberta, Canada, from 1976 to 2012 and in Charadriiformes (shorebirds and gulls) at Delaware Bay in New Jersey and Delaware, USA, from 1985 to 2012 provides a large dataset that has contributed to the general understanding of influenza in the reservoir species and has established some of the ecological principles of influenza in nature,Citation3,Citation4,Citation5 but these studies have not been evaluated for their predictive value. Of the 16 hemagglutinin (HA) subtypes of IAVs known to be circulating in the aquatic bird reservoir, two – H5 and H7 – have the unique ability to become highly pathogenic (HP) in domestic gallinaceous poultry.Citation6,Citation7
Between 2002 and the present there have been four outbreaks of HP H7N3 influenza in poultry in the Americas, as represented by A/Chicken/Chile/4322/2002,Citation8 A/Chicken/Canada/AVF2/2004,Citation9 A/Chicken/Saskatchewan/HR00011/2007,Citation10 and A/Chicken/Jalisco/CPA1/2012.Citation11 Each of these outbreaks of HP H7N3 IAVs caused high mortality in gallinaceous poultry. These viruses acquired additional basic amino acids juxtaposed to the cleavage site in the HA, which is necessary for the HP phenotype. Generally HPAI HA gene segments acquire these basic amino acids through insertion of short stretches of adenosine and guanosine (e.g., AGAAAAAAAAGA) nucleotides and if these insertions are in frame, they will be translated to lysine/arginine. In one case, it appears that these basic residues were acquired by non-homologous recombination.Citation12 The molecular mechanism by which HPAI typically evolves has not been clearly delineated, and we hypothesized that specific polymerase gene segments/constellations may favor the evolution of HPAI. Additionally, the HP H7N3 IAV from chickens in British Columbia in 2004 was associated with mild respiratory disease and unilateral conjunctivitis in two humans,Citation13 and the Mexican HP H7N3 from 2012 was isolated from conjunctivitis in humans.Citation14
Multiple outbreaks of low pathogenic (LP) H7N2 influenza occurred in chickens and turkeys between 1996 and 2004 in the eastern United States from South Carolina to Massachusetts, including Pennsylvania, New Jersey, and Ohio (reviewed in the ref. 15). In turkeys, the H7N2 viruses caused respiratory disease, egg production drop, lethargy, and depression,Citation16 but there was usually limited mortality. In chickens, the H7N2 viruses were characterized by rapid spread and multi-causal respiratory disease.Citation17 During 1996–2004, H7N2 IAVs were frequently isolated from live-bird markets along the east coast of the United States, which contributed to their spread and evolution. The spread and evolution of H7 viruses also lead to infrequent zoonosis such as: isolation of a LP H7N2 virus from the respiratory tract of an immunocompromised patient with mild respiratory disease;Citation18 the isolation of a LP H7N7 IAV from Phoca vitulina (harbor seals) from a human with conjunctivitis after an experimentally infected seal sneezed in the face of the handler.Citation19
Understanding the evolution of LP and HP H7 IAVs is important to animal health and pandemic preparedness. Because LP H7N9 influenza viruses have recently emerged in humans in China and have caused high mortalityCitation20 we determined the presence of H7N9 in wild aquatic birds and domestic poultry in America. In addition, because HP H7 influenza viruses have emerged multiple times in poultry in the Americas and have occasionally transmitted to humans and LP H7N2 viruses have spread in domestic poultry for several years in the United States, we examined our long-term surveillance data to evaluate its value as a predictive tool to forecast H7 outbreaks and gain a better understanding of the emergence of HP IAV. The present study utilizes long-term surveillance data on influenza in migratory birds to determine: (i) the patterns of point prevalence of H7 influenza in wild birds in the Americas, (ii) which HA–neuraminidase (NA) combinations have predominated, (iii) whether Anseriformes (ducks) and Charadriiformes (shorebirds) have similar or different HA-NA combinations, (iv) if there is concordance between peaks of prevalence and transmission of H7 in wild aquatic birds and the emergence of H7 IAVs in poultry and people, and (v) if a particular lineage of RNA polymerase gene segments/constellations have repeatedly given rise to HPAI viruses. Phylogenetic and genomic constellation analyses were used to infer the precursors of the HP influenza outbreaks and their estimated dates of emergence, as well as to determine whether HP viruses emerge in the context of specific internal gene segment sequences.
Material and methods
Surveillance sites and sampling
Wild ducks. Surveillance was conducted at various lakes throughout Alberta, Canada at various timepoints spanning July through August from 1976 to 2012. Cloacal or paired cloacal and oropharyngeal swabs from hatch-year and after hatch-year ducks were collected from birds at pre-migration staging areas. Details about collection sites, species sampled, sample collection procedures, and transportation of specimens have been reported.Citation1,Citation21 A total 3693 IAVs were isolated from 17 369 samples (21.3%) representing 15 989 birds (3575 influenza positive birds (22.4%).
Shorebirds. Fecal samples, along with a limited number of cloacal swabs, were collected from migrating shorebirds at their stopover site at Delaware Bay, USA during May from 1985 to 2012. Fecal samples from gull species were also obtained. Samples came primarily from beaches in New Jersey, but from 1985 to 1990, a few beaches in Delaware also served as collection sites. Details about collection sites, species sampled, sample collection procedures, and transportation of specimens have been reported previously.Citation1,Citation22 A total of 1085 IAVs were isolated from 10 430 samples (10.4%).
Isolation and identification procedures
Viruses were isolated in 10-day-old embryonated hen’s eggs according to the protocol of Palmer et al. (1975).Citation23 The HA and NA subtypes of the virus isolates were identified by hemagglutination inhibition (HAI)Citation23 and neuraminidase inhibition (NAI)Citation24 assays, respectively. In instances where the subtype could not be determined by HAI or NAI, we employed reverse transcription-polymerase chain reaction (RT-PCR) to amplify the HA and/or NA gene,Citation25 followed by nucleotide sequencing and BLAST comparisons with GenBank records.
Survey of LP H7 in domestic poultry
Information on the detection of H7 avian influenza in domestic poultry in the Americas between 1976 and 2013 was obtained from reports to the World Animal Health Association for the years 2005–2013,Citation26 reports to the US Animal Health Association for the years 1976–2013 (Supplementary Table S1) and reports made at the International Symposia on Avian Influenza for years 1986–2005.Citation27,Citation28,Citation29,Citation30
Statistical analysis
A chi-square test was used to compare the frequency of detection of H7N3 versus any other H7-NA combination. This test was also used to compare the frequency of detection of any H7 subtype or H7N3 between ducks and shorebirds. PASW (SPSS) 18 software was used (IBM, Armonk, NY).
Viral sequencing
Sequencing and genome assembly for the majority of viruses was done using a next-generation sequencing (NGS) pipeline at J. Craig Venter Institute that used the Roche 454 GS-FLX and the Illumina HiSeq 2000. Viral RNA was isolated and subjected to multi-segment RT-PCR (M-RTPCR),Citation31,Citation32 which simultaneously and specifically amplifies all IAV segments in a single reaction, irrespective of the virus subtype. The amplicons for each sample were primed with barcoded random hexamers using our modified sequence independent single primer amplification protocol,Citation33,Citation34 amplified, and size-selected using agarose gel electrophoresis. NGS libraries were subsequently prepared for sequencing on either the Roche 454 GS-FLX platform using Titanium chemistry or the Illumina HiSeq 2000 (2× 100 bp) and/or MiSeq (2× 250 bp). The sequence reads were sorted by barcode, trimmed, searched by TBLASTX against custom nucleotide databases of full-length influenza A segments downloaded from GenBank to filter out both chimeric influenza sequences and non-influenza sequences amplified during the random-hexamer-primed amplification. The filtered GS-FLX reads were then binned by segment and de novo assembled using CLC Bio’s clc_novo_assemble program. The resulting contigs were searched against the corresponding custom full-length influenza segment nucleotide database to find the closest reference sequence for each segment. Both GS-FLX and Illumina reads were then used for reference-based assembly using CLC Bio’s clc_ref_assemble_long program.
Phylogenetic and constellation analyses
Sequence collection and curation. All complete avian and human H7 genomes isolated in North and South America were downloaded from GenBank, including historical references and equine H7N7 genomes. We also included South American H7 viruses known to have produced a HP outbreak whose genomes were not fully sequenced. Because our initial dataset included duplicated genomes due to variable strain names, we deduplicated the dataset by standardizing case, spelling, and abbreviations in the strain names for all segments. This yielded 467 genomes for the initial phylogenetic and constellation analyses.
Maximum likelihood (ML) analysis. Multiple alignment using fast Fourier transform (MAFFT) v7Citation35,Citation36,Citation37 was used to construct an HA alignment, which was checked and trimmed by hand to the coding region. jModelTest 2.4Citation38 was used to determine that the most appropriate nucleotide substitution model for our HA data was a general time reversible (GTR) model with a gamma rate distribution and invariant sites (GTR-IG). We performed a ML phylogenetic analysis for the HA nucleotide sequences using the GARLI 2.0 web service.Citation39,Citation40 The resultant tree was colored based on the taxonomic Order of the host species (e.g., Anseriformes, Galliformes, etc.) using in-house PERL scripts.
Bayesian analysis. Using our initial ML and constellation analysis results, we chose a subset of our total genomes for a Bayesian analysis of HA nucleotide sequences by selecting viral strains with unique phylogenetic and reassortant histories while ensuring that host diversity, available NA subtypes, and historical reference strains were retained. To determine if our subsampled data exhibited temporal qualities, we performed an exploratory analysis with Path-O-Gen (available at http://tree.bio.ed.ac.uk/software/pathogen/) to measure root-to-tip divergence for a subsampled HA ML tree constructed using MEGA6Citation41 with a GTR-IG substitution model. The results supported the use of a molecular clock model, so we proceeded with our time-based Bayesian analysis using BEAST v1.8Citation42,Citation43 on the Cyber Infrastructure for Phylogenetic Research Science Gateway (available at http://www.phylo.org/).Citation44,Citation45 Using a Bayes factor test to compare the use of a strict versus lognormal relaxed molecular clock for our dataset, we determined that a lognormal relaxed clock best fit our data. Tips were dated by year and Markov Chain Monte Carlo chains were run with a length of 100 million using the substitution model determined above; parameters and trees were sampled every 10 000 iterations. The skygrid coalescent model was used with 50 partitions over 50 years. Default priors were used, with the exception that the Continuous-time Markov Chain rate reference was used for the uncorrelated relaxed clock mean. All analyses were evaluated with Tracer v1.6 to evaluate the success of the chain sampling based on effective sample size values for each parameter, and additional chains were run as needed. We constructed a maximum clade credibility tree for HA using TreeAnnotator. The tree was colored by taxonomic Order as described above.
Genome constellation analysis. Genomic variation and reassortment was assessed using a novel genome constellation analysis technique (Seth A Schobel et al., unpublished data). For each segment, sequences were aligned using MAFFT v7 and clustered using the ANDES suite of deep sequence analysis software.Citation46 Each multiple sequence alignment was converted to a distance matrix and clustered using the complete linkage algorithm, which optimizes clusters by merging clusters with the smallest distance between two clusters as measured by their respective farthest members. Distance is measured by sequence identity, allowing the resultant dendrograms to be cut/clustered at any desired percent identity threshold and guaranteeing that all members of a cluster are within the specified percent identity cutoff. Gene cluster assignments were made for each segment using the same cutoff and combined into a genome constellation, which was visualized using custom software called OrionPlot.Citation47 The cutoff for this analysis was selected based on biological relevance and ease of visualization for the amount of diversity in our dataset.
Compliances
Banding of ducks in Alberta, Canada was conducted in accordance with the Environment Canada scientific permit (number 10810) to capture and band migratory birds. All work with virus isolates was performed in US Department of Agriculture (USDA)-inspected and approved Biosafety Level 2 (BSL2)-enhanced laboratories, and all procedures involving infectious viruses were reviewed and approved by the St. Jude Children’s Research Hospital Institutional Biosafety Committee.
Results
Distribution of H7 influenza viruses and HA-NA subtype diversity in wild aquatic birds at North American surveillance sites
Long-term surveillance of IAVs in migratory ducks in Alberta, Canada, from 1976 to 2012 and in shorebirds and gulls at Delaware Bay, United States, from 1985 to 2012 reveals differences in the frequency of NA subtypes that have been detected in combination with the H7 HA (). There was a statistically significant (P value < 0.001) predominance of H7N3 over all other H7-NA combinations in the sampled species (29 duck virus isolates and 44 shorebird virus isolates). The H7 subtype was more likely to occur in shorebirds and gulls (6.18%) rather than ducks (1.11%) (P value < 0.001). Similarly, H7N3 was more frequently detected in shorebirds and gulls (4.06%) than ducks (0.79%) (P value < 0.001). In ducks, N8 was the next most frequent NA subtype (four virus isolates), followed by N1 (three virus isolates), N5 and N9 (two virus isolates each), and N2 (one virus isolate). In ducks, N4, N6, and N7 were noticeably absent. In shorebirds, eight of the nine NA subtypes found in wild birds were detected in combination with an H7 HA; the most frequent NA subtype after N3 was N7 (seven virus isolates), followed by N4 (five virus isolates), N2 (four virus isolates), N5 (three virus isolates), N8 (two virus isolates), and N1 and N9 (one virus isolate each). As was the case for ducks, N6 was not detected in shorebirds.
Table 1 Influenza H7-NA subtypes isolated during surveillance studies in wild aquatic birds in Alberta, Canada, and Delaware Bay, USA, between 1976 and 2012
Temporal distribution of the H7N3 subtype in aquatic birds at North American surveillance sites
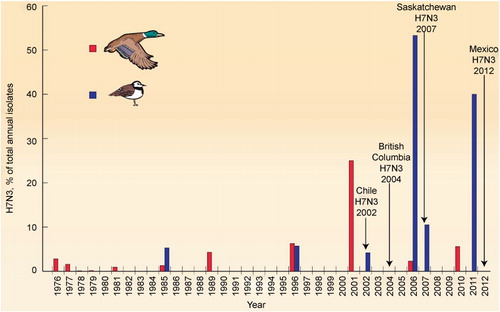
Of all H7-NA combinations in both migratory ducks and shorebirds, H7N3 was the most common. The H7N3 combination occurred 12 times in ducks over 36 years of surveillance and seven times in shorebirds over 27 years of surveillance. H7N3 was detected much more frequently than any other H7-NA combination, sometimes occurring in both ducks and shorebirds within the same year (e.g., 1985 and 1996) or in consecutive years (e.g., 2001/2002 and 2010/2011) (). Since 2000, there have been three major peaks of H7N3 activity: one in ducks in 2001 that contributed 25% of the total isolates for the year, one in shorebirds in 2006–2007 that constituted 55% of the total isolates those years, and another in 2011 in shorebirds that made up 40% of the total isolates that year.
Reports of LP H7 IAVs in domestic poultry in the Americas
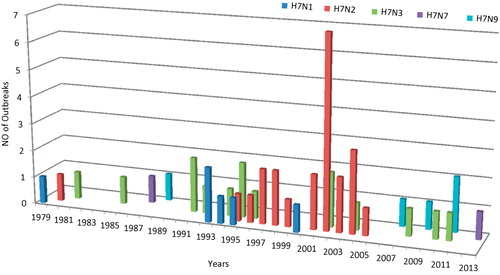
To determine the incidence of H7 IAVs in domestic poultry in the United States from 1976 to 2013 (), we used USDA reports from live bird market influenza surveillance,Citation27,Citation28,Citation29,Citation30 reports of LP viruses on domestic poultry farms from the World Organisation for Animal Health (OIE),Citation26 and reports from the United States Animal Health Association (Supplementary Table S1). H7N2 was detected most frequently, with 17 reports from markets and 22 reports from poultry farms; H7N3 was the next most frequently reported subtype (2 market outbreaks, 14 farm outbreaks), followed by H7N9 (6 farm outbreaks). The frequency of isolation of H7N1 and H7N7 was surprisingly lower, with only four and two reports, respectively. It is notable that four NA subtypes (N4, N5, N6, N8) were not detected in combination with H7 in domestic poultry, and two of these subtypes were not isolated from ducks (N4 and N6) or shorebirds and gulls (N6) in our surveillance studies.
Table 2 Influenza H7-NA subtypes reported in domestic poultry in the Americas between 1976 and 2013
It is noteworthy that LP H7N9 was the third most frequently detected H7–NA combination reported in commercial poultry (). These H7N9-associated outbreaks of respiratory disease in poultry occurred in Nebraska turkeys in 2007, Minnesota turkeys in 2009 and 2011, and Kentucky chickens in 2009; in total, these outbreaks involved over 300 000 birds.Citation26 However, H7N9 was detected in only three sampling periods during our long-term surveillance in wild ducks and shorebirds: two duck isolates from 1999 and 2004 and one shorebird isolate from 1995. It was not detected in live bird markets from 1979 to 2013.
Since 2003, regulatory changes by the USDA have mandated the culling of all LP, as well as HP, H5, and H7 viruses. Thus, the LP H7N9 outbreaks of influenza in domestic poultry were stamped out. The timeline for the introductions of LP H7N2, H7N3, and H7N9 into domestic poultry is shown in .
Reports of HP H7 IAVs in the Americas
Since the late 1950s, the only HP H7 IAV detected in the Americas has been H7N3.Citation15 Prior to that time, HP IAVs were characterized based on disease signs in poultry and collectively referred to as “fowl plague”; the H5 and H7 subtypes were not specified, and the NA was not characterized. The emergence of a single HP subtype, H7N3, in the Americas contrasts with Eurasia and Australasia where multiple HP subtypes – H7N1, H7N3, H7N4, and H7N7 – have been sporadically reported in domestic poultry.Citation15 In the Americas, HP H7N3 emerged in chickens in Chile in 2002 (A/Chicken/Chile/4322/2002); in chickens in British Columbia, Canada in 2004 (A/Chicken/Canada/AVF2/2004); in chickens in Saskatchewan, Canada in 2007 (A/Chicken/Saskatchewan/HR00011/2007); and in chickens in Jalisco, Mexico in 2012 (A/Chicken/Jalisco/CPA1/2012) (). There was general concordance between the major peaks of LP H7N3 influenza detected in wild birds and outbreaks of HP H7N3 in domestic gallinaceous poultry.
Gene flow from wild to domestic birds in the Americas
Phylogenetic analysis of the H7 gene from both LP and HP viruses from wild and domestic avian species revealed that the H7 genes of viruses isolated from wild ducks and shorebirds are closely related to the LP and HP H7 genes of viruses isolated from North and South American poultry ( and Supplementary Figure S1). The four HP poultry outbreaks in the Americas arose from polyphyletic strains (the green and red strain names within red boxes in ), indicating that each HP outbreak was an independent emergence event. Therefore, each event represents a separate introduction of LP avian influenza into poultry from the wild aquatic bird reservoir, most likely from dabbling ducks. H7 sequences from each H7N3 domestic poultry outbreak are most closely related to H7 sequences isolated from wild ducks during the same year as the outbreak and/or in the years immediately preceding the outbreak (dark blue strain names in representing Anseriformes). An alignment of the HP HA from each outbreak indicated that the polybasic cleavage site sequence varied among the four outbreak strains (), further supporting their independent origins. The South American LP and HP strains form a distinct H7 lineage from the North American strains, which diverged around 1955 (with a 95% highest posterior density (HPD) range from 1938 to 1969) (). All modern American H7 lineages diverged from historic European H7 strains around 1877 (95% HPD range from 1836 to 1911).
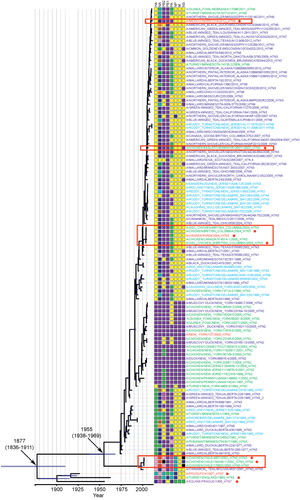

A genomic constellation analysis where each segment was clustered at a 90% nucleotide identity cutoff demonstrated that the HP H7 segments emerged in the context of different internal gene segment combinations (). The three HP H7N3 lineages that emerged in North America possessed similar HA, NA, PB2, PB1, NP, and M genes at a 90% nucleotide identity cutoff, but they differed in their PA and NS genes, while the 2002 HP outbreak in Chile was generated by precursor viruses distinct in most gene segments from LP avian influenza strains found in North America. Using a more stringent evolutionary cutoff (95% nucleotide identity) (), each HP H7N3 outbreak in domestic poultry and human spillover contains almost fully unique genome constellations, as evidenced by the different colors for strains from each outbreak within a column that represents one of the eight gene segments (). The only exceptions are the 2012 Mexican and 2004 British Columbian outbreaks that had NS segments that share 95% nucleotide identity, M segments that are within 95% nucleotide identity for all three North American outbreaks, and the 2007 Saskatchewan and 2012 Mexican outbreaks that had PB1 segments that share 95% identity. When multiple sequences are available from a single outbreak, their genome constellations are identical at 95% nucleotide identity. Comparison of wild bird sequences that shared the closest cophenetic distance to the H7N3 poultry and human outbreak sequences in the H7 Bayesian phylogeny showed that the duck HA, PB1, and M sequences usually clustered with the poultry and human outbreak sequences at 95% nucleotide identity. In addition, when N3 strains were available for comparison, the NA duck sequences also clustered with the poultry and human outbreak strains at 95% nucleotide identity. The remaining segments usually differed between the most cophenetic wild bird sequences and the poultry and human outbreak sequences, demonstrating that those wild bird segments were not closely related to the outbreak sequences. This is likely a reflection of the fact that wild bird sampling for influenza surveillance is not performed at a resolution across the Americas that allows for the identification of the matching precursor wild bird strains that emerge in poultry. Collectively, the genomic constellation analysis strongly suggests that gene segments/viruses flow from wild birds to domestic poultry and demonstrates that HP H7 poultry outbreaks represent independent emergence events arising from different polymerase gene lineages.
The New York live bird market H7N2 isolates collected in the 2000s form their own distinct HA phylogenetic lineage ( and Supplementary Figure S1), with no clear origin from the North American wild bird populations that were sampled in this study. This is also evidenced by these viruses belonging to their own unique HA cluster at a 90% nucleotide identity cutoff (, purple boxes in the first column of the genome constellations).
Discussion
The available evidence on the emergence of HP avian IAVs supports the hypothesis that they emerge from a reservoir of LP IAVs in aquatic birds. Transmission of viruses between wild aquatic birds and domestic poultry may occur in a number of ways. When migratory birds scavenge food, there is direct contact between migratory waterfowl and free-range domestic species including ducks, geese, and “backyard” poultry. Additionally, there is indirect contact between migratory waterfowl and domestic poultry through contaminated water supplies because the majority of the IAVs in migratory waterfowl replicate in the intestinal tract and are transmitted by fecal–oral transmission through environmental contamination, which includes contaminated, untreated water.
After introduction into domestic poultry from wild migratory birds, IAVs are spread by humans either directly by carrying fomites or through the poultry trade, as birds are moved from hatcheries to poultry farms. Additionally, live poultry markets are an optimal place to amplify and spread IAVs, and the number of such markets in the major cities on both coasts of the United States is higher than most people appreciate. Live bird markets have been described as the breeding grounds for IAVsCitation48 and are an optimal site for surveillance for IAVs in domestic poultry.
The emergence of IAVs in domestic avian species and the associated transmissions to mammals, including swine and humans, are currently unpredictable. Of current concern are the H7 IAVs, particularly with H7N3 IAVs repeatedly becoming HP for poultry in the Americas and occasionally spilling over into humans. In addition, LP H7N9 viruses in poultry have emerged in China and are HP in humans.Citation20 Here, we demonstrate that long-term surveillance for influenza in aquatic bird reservoir species can serve as an early warning system for determining when LP and HP poultry outbreaks are likely to occur for certain subtypes. While there was general concordance between the emergence of HP H7N3 with peaks of virus prevalence in shorebirds and ducks, there were no predictive peaks of activity in wild birds for either LP H7N2 or H7N9 in domestic poultry. For H7N3, there were multiple independent introductions into domestic poultry that may have occurred by direct or indirect contact between wild birds and free-ranging poultry. While the Chilean and Canadian HP H7N3 outbreaks were stamped out, the Mexican outbreak was not initially eradicated and spread widely in poultry in seven states in Mexico, sometimes causing conjunctivitis in humans. Adoption of poultry vaccinations has reduced the severity of disease in chickens in Mexico but has not reduced transmission. There is also concern that transmission to humans may be more frequent than reported.
Overall for wild bird H7 IAVs, shorebirds carried the majority of possible HA–NA combinations; eight of the nine NA subtypes found in aquatic birds were detected in shorebirds, while only six of the nine NA subtypes were found in ducks. The most frequent combination was H7N3, which was found in both shorebird and duck species, but more frequently in shorebirds. However, our genomic analyses suggest that H7 poultry strains in the Americas are most closely related to H7 strains in ducks, rather than shorebirds. This suggests that dabbling ducks are the primary source of the H7 domestic poultry viruses, and broadening surveillance to additional sites would provide finer resolution for the evolution and transmission dynamics of H7 gene flow between shorebirds, ducks, and domestic poultry.
Our surveillance studies in shorebirds were conducted each May when the birds stopover in Delaware Bay to refuel on horseshoe crab eggs en route to their breeding grounds in Northern Canada. At this time, the shorebirds have migrated directly from South America and are shedding high levels of IAVs of most subtype combinations. Delaware Bay has been recognized as a “hot spot” for IAV isolation.Citation4 Interestingly, all H7 viruses detected in these Delaware Bay shorebirds match North American strains and are phylogenetically distinct from available poultry and duck South American H7 strains. However, no H7 sequences from South American shorebirds were available for comparison. In the future, surveillance and genomic sequencing from South American wild bird species is important to understand long-range intercontinental transmission dynamics of H7 viruses.
There appears to have been a single introduction of H7N2 from its wild reservoir into domestic poultry in 1994, and this subtype has since been maintained in live poultry markets and poultry houses in the mid-Atlantic and Northeastern United States. The detection of multiple H7N9 viruses in domestic poultry (primarily turkeys), and only three H7N9 viruses detected during 36 years of wild bird surveillance, suggests that these viruses are rarely sampled in the current surveillance or were generated within poultry and/or have a preference for domestic poultry. These H7N9 viruses probably arose through direct contact between free-range reared birds and wild birds. Stamping-out protocols presumably eliminated the H7N9 viruses from domestic turkeys and chickens, and the virus did not enter the live poultry market system.
While the HP phenotype is a polygenic property, the HA is considered the most critical determinant because cleavage activation of the polybasic HA1/HA2 cleavage site by ubiquitous cellular protease(s) leads to systemic spread. There appears to be two molecular mechanisms by which HP viruses are generated from LP precursors. One mechanism involves stuttering of the polymerase during RNA replication,Citation49 whereas an atypical mechanism involved the insertion of a series of basic amino acids in the connecting peptide of the HA via insertion of a fragment of another viral RNA.Citation12 The genomic analysis conducted illustrates that HP H7 poultry outbreaks represent independent emergence of HP HA cleavage site acquisition events, which arose within the context of different polymerase gene segment lineages. One might anticipate that the RNA polymerase gene constellations that give rise to stuttering versus recombination mediated HP HAs would stem from different lineages. However, it also appears the multiple HP genotypes generated via stuttering-like mechanism are also generated from different RNA polymerase gene segment lineages.
Although surveillance of influenza in wild aquatic birds has provided information on the ecology and origins of pandemic IAVs, its overall predictive value remains limited to specific subtypes. Our surveillance indicates that for H7N3 viruses there are correlates between peaks of activity in wild birds and outbreaks of HP influenza in domestic poultry. While it appears that H7N3 activity in wild aquatic birds is a precursor for outbreaks in poultry, this could not be validated statistically in our study due to a small number of occurrences and potential unreported outbreaks. However, forewarning of the circulation of H7N3 IAV in wild birds and increased biosecurity on poultry farms could potentially prevent the emergence of HP avian influenza outbreaks. The detection and spread of LP H5 or H7 in gallinaceous poultry is considered indicative of the emergence of a HP strain and is therefore stamped out, typically by depopulation. Changes in turkey husbandry, from open range to indoor production, and the culling of all LP and HP H5- or H7-infected birds have already contributed to reducing the emergence of HP influenza in the Americas. Early warning of potential HP H5 or H7 outbreaks, at least in the Americas, may be possible using wild bird influenza surveillance, especially at hot spots on each flyway.
Supplementary Table S1
Download MS Word (16.1 KB)Supplementary Figure S1
Download PDF (52 MB)The authors thank the JCVI team of researchers that provided technical expertise in support of the viral sequencing and assembly pipeline for this project, especially Rebecca A Halpin, Nadia Fedorova, and Susmita Shrivastava. This project was funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, USA under contract numbers HHSN272200900007C (Genome Sequencing Center for Infectious Diseases), HHSN266200700005C and HHSN2720140006C, and by American Lebanese Syrian Associated Charities. The data for this manuscript and its preparation were generated while David E Wentworth was employed at JCVI. The opinions expressed in this article are the authors’ own and do not reflect the views of the Centers for Disease Control, the Department of Health and Human Services, or the United States Government.
Supplementary Information for this article can be found on the Emerging Microbes & Infections website (http://www.nature.com/emi)
Related Research Data
- Krauss S, Walker D, Pryor SP et al.Influenza A viruses of migrating wild aquatic birds in North America. Vector Borne Zoonotic Dis 2004; 4: 177–189.
- Munster VJ, Baas C, Lexmond P et al.Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog 2007; 3: e61.
- Krauss S, Obert CA, Franks J et al.Influenza in migratory birds and evidence of limited intercontinental virus exchange. PLoS Pathog 2007; 3: e167.
- Krauss S, Stallknecht DE, Negovetich NJ, Niles LJ, Webby RJ, Webster RG.Coincident ruddy turnstone migration and horseshoe crab spawning creates an ecological ‘hot spot’ for influenza viruses. Proc Biol Sci 2010; 277: 3373–3379.
- Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y.Evolution and ecology of influenza A viruses. Microbiol Rev 1992; 56: 152–179.
- Kaleta EF, Rulke CPA.The beginning and spread of fowl plague (H7 High Pathogenicity Avian Influenza) across Europe and Asia (1878-1955).In: Swayne DE (ed). Avian influenza.1st ed. Ames, IA: Blackwell, 2008: 145–189.
- Rott R, Schäfer W.[Physico-chemical and biological characters of Virus N and its relations to the influenza sub-group A of myxoviruses.] Zentralblatt für Veterinärmedizin 1960; 7: 237–248.German.
- Rojas H, Moreira R, Avalos P, Capua I, Marangon S.Avian influenza in poultry in Chile. Vet Rec 2002; 151: 188.
- Hirst M, Astell CR, Griffith M et al.Novel avian influenza H7N3 strain outbreak, British Columbia. Emerg Infect Dis 2004; 10: 2192–2195.
- Berhane Y, Hisanaga T, Kehler H et al.Highly pathogenic avian influenza virus A (H7N3) in domestic poultry, Saskatchewan, Canada, 2007. Emerg Infect Dis 2009; 15: 1492–1495.
- Maurer-Stroh S, Lee RT, Gunalan V, Eisenhaber F.The highly pathogenic H7N3 avian influenza strain from July 2012 in Mexico acquired an extended cleavage site through recombination with host 28S rRNA. Virol J 2013; 10: 139.
- Suarez DL, Senne DA, Banks J et al.Recombination resulting in virulence shift in avian influenza outbreak, Chile. Emerg Infect Dis 2004; 10: 693–699.
- Tweed SA, Skowronski DM, David ST et al.Human illness from avian influenza H7N3, British Columbia. Emerg Infect Dis 2004; 10: 2196–2199.
- Belser JA, Davis CT, Balish A et al.Pathogenesis, transmissibility, and ocular tropism of a highly pathogenic avian influenza A (H7N3) virus associated with human conjunctivitis. J Virol 2013; 87: 5746–5754.
- Swayne DE.The global nature of avian influenza.In: Swayne DE (ed). Avian influenza.1st ed. Ames, IA: Blackwell, 2008: 123–143.
- Davison S, Eckroade RJ, Ziegler AF.A review of the 1996-98 nonpathogenic H7N2 avian influenza outbreak in Pennsylvania. Avian Dis 2003; 47: 823–827.
- Dunn PA, Wallner-Pendleton EA, Lu H et al.Summary of the 2001-02 Pennsylvania H7N2 low pathogenicity avian influenza outbreak in meat type chickens. Avian Dis 2003; 47: 812–816.
- Ostrowsky B, Huang A, Terry W et al.Low pathogenic avian influenza A (H7N2) virus infection in immunocompromised adult, New York, USA, 2003. Emerg Infect Dis 2012; 18: 1128–1131.
- Webster RG, Geraci J, Petursson G, Skirnisson K.Conjunctivitis in human beings caused by influenza A virus of seals. N Engl J Med 1981; 304: 911.
- Gao R, Cao B, Hu Y et al.Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 2013; 368: 1888–1897.
- Sharp GB, Kawaoka Y, Wright SM, Turner B, Hinshaw V, Webster RG.Wild ducks are the reservoir for only a limited number of influenza A subtypes. Epidemiol Infect 1993; 110: 161–176.
- Kawaoka Y, Chambers TM, Sladen WL, Webster RG.Is the gene pool of influenza viruses in shorebirds and gulls different from that in wild ducks? Virology 1988; 163: 247–250.
- Palmer DF. Advanced laboratory techniques for influenza diagnosis.Atlanta, GA: Center for Disease Control, 1975.
- Aymard-Henry M, Coleman MT, Dowdle WR, Laver WG, Schild GC, Webster RG.Influenzavirus neuraminidase and neuraminidase-inhibition test procedures. Bull World Health Organ 1973; 48: 199–202.
- Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR.Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 2001; 146: 2275–2289.
- World Organisation for Animal Health (OIE). World Animal Health Information Database (WAHID) interface for low pathogenic avian influenza (poultry).Paris: OIE, 2013.Available at http://www.oie.int/wahis_2/public/wahid.php/Diseaseinformation/Immsummary (accessed 3 December 2014).
- Pearson JE, Senne DA, Panigrahy B. Avian influenza in the Western Hemisphere including the Pacific Basin 1992-1996.Jacksonville: American Association of Avian Pathologists, 2003.Available at http://www.jstor.org/stable/3298794 (accessed 2 October 2014).
- Pearson JE, Senne DA, Panigrahy B. Avian Influenza in the Western Hemisphere including the Pacific Basin 1986-1992.Jacksonville: American Association of Avian Pathologists, 2003.Available at http://www.jstor.org/stable/3298666 (accessed 2 October 2014).
- Senne DA.Avian Influenza in the Western Hemisphere Including the Pacific Islands and Australia. Avian Dis 2003; 47: 798–805.
- Senne DA.Avian Influenza in North and South America, 2002-2005 (Influenza aviar en Norte y Suramérica, 2002-2005). Avian Dis 2007; 51: 167–173.
- Zhou B, Donnelly ME, Scholes DT et al.Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol 2009; 83: 10309–10313.
- Zhou B, Wentworth DE.Influenza A virus molecular virology techniques. Methods Mol Biol 2012; 865: 175–192.
- Djikeng A, Halpin R, Kuzmickas R et al.Viral genome sequencing by random priming methods. BMC Genomics 2008; 9: 5.
- Djikeng A, Spiro D.Advancing full length genome sequencing for human RNA viral pathogens. Future Virol 2009; 4: 47–53.
- Katoh K, Misawa K, Kuma K, Miyata T.MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002; 30: 3059–3066.
- Katoh K, Standley DM.MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013; 30: 772–780.
- Katoh K, Toh H.Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 2008; 9: 286–298.
- Darriba D, Taboada GL, Doallo R, Posada D.jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 2012; 9: 772.
- Bazinet AL, Cummings MP.The Lattice Project: a grid research and production environment combining multiple grid computing models.In: Weber MHW (ed) Distributed & grid computing – science made transparent for everyone. Principles, applications and supporting communities.Marburg: Rechenkraft.net, 2008: 2–13.
- Zwickl DJ.Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion.PhD thesis, The University of Texas at Austin, Austin, TX, 2006.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S.MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 2013; 30: 2725–2729.
- Drummond AJ, Rambaut A.BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 2007; 7: 214.
- Drummond AJ, Suchard MA, Xie D, Rambaut A.Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 2012; 29: 1969–1973.
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees.Gateway Computing Environments Workshop (GCE); 14 November 2010; New Orleans, LA, USA.
- Miller MA, Pfeiffer W, Schwartz T.The CIPRES science gateway: a community resource for phylogenetic analyses. Proceedings of the 2011 TeraGrid Conference: Extreme Digital Discovery; 18–21 July 2011; Salt Lake City, Utah, USA.
- Li K, Venter E, Yooseph S et al.ANDES: Statistical tools for the ANalyses of DEep Sequencing. BMC Res Notes 2010; 3: 199.
- Schobel-McHugh R. OrionPlot.San Francisco, CA: GitHit, 2014.Available at https://github.com/sschobel/orion-plot/tree/master/OrionPlot/dist/alpha (accessed 2 October 2014).
- Senne DA, Suarez DL, Pedersen JC, Panigrahy B.Molecular and biological characteristics of H5 and H7 avian influenza viruses in live-bird markets of the northeastern United States, 1994-2001. Avian Dis 2003; 47: 898–904.
- Perdue M, Crawford J, Garcia M, Latimer J, Swayne D. Occurrence and possible mechanisms of cleavage-site insertions in the avian influenza hemagglutinin gene.Jacksonville: American Association of Avian Pathologists, 2003.Available at http://www.jstor.org/stable/3298820 (accessed 2 October 2014).