
Abstract
Less than 20 sporadic cases of human Zika virus (ZIKV) infection were reported in Africa and Asia before 2007, but large outbreaks involving up to 73% of the populations on the Pacific islands have started since 2007, and spread to the Americas in 2014. Moreover, the clinical manifestation of ZIKV infection has apparently changed, as evident by increasing reports of neurological complications, such as Guillain–Barré syndrome in adults and congenital anomalies in neonates. We comprehensively compared the genome sequences of pre-epidemic and epidemic ZIKV strains with complete genome or complete polyprotein sequences available in GenBank. Besides the reported phylogenetic clustering of the epidemic strains with the Asian lineage, we found that the topology of phylogenetic tree of all coding regions is the same except that of the non-structural 2B (NS2B) coding region. This finding was confirmed by bootscan analysis and multiple sequence alignment, which suggested the presence of a fragment of genetic recombination at NS2B with that of Spondweni virus. Moreover, the representative epidemic strain possesses one large bulge of nine bases instead of an external loop on the first stem-loop structure at the 3′-untranslated region just distal to the stop codon of the NS5 in the 1947 pre-epidemic prototype strain. Fifteen amino acid substitutions are found in the epidemic strains when compared with the pre-epidemic strains. As mutations in other flaviviruses can be associated with changes in virulence, replication efficiency, antigenic epitopes and host tropism, further studies would be important to ascertain the biological significance of these genomic changes.
Emerging Microbes and Infections (2016) 5, e22; doi:10.1038/emi.2016.48; published online 16 March 2016
INTRODUCTION
Zika virus (ZIKV) is an emerging mosquito-borne human-pathogenic flavivirus that has been mostly neglected due to its mild clinical manifestations and limited spread in restricted geographical regions in the first 60 years after its discovery.Citation1 ZIKV was first isolated from the serum of a febrile sentinel rhesus macaque in 1947 in Zika Forest of Uganda.Citation2 Between 1947 and 2006, <20 cases of human ZIKV infection were reported in the literature.Citation1 These cases were geographically restricted in certain African (African lineage) and Southeast Asian (Asian lineage) countries. The first documented sizable outbreak of human ZIKV infection outside Africa and Asia occurred on Yap Island of the French States of Micronesia in 2007, during which 73% of the Yap population became infected.Citation3 ZIKV then spread to other Pacific islands, and arrived in the western hemisphere in 2014 (Easter Island, Chile).Citation4, Citation5, Citation6 Since then, many countries in the Americas have reported autochthonous cases of ZIKV infection. Brazil alone has reported an estimated 500 000–1 500 000 human cases of ZIKV infection in 2015.Citation7 Although most patients with ZIKV infection are asymptomatic or have mild symptoms, life-threatening complications such as Guillain–Barré syndrome, thrombocytopenic purpura, and fatal disseminated disease in immunosuppressed hosts have been reported.Citation1, Citation3, Citation8 Furthermore, preliminary epidemiological and virological data suggest that congenital ZIKV infection may be associated with microcephaly and other congenital anomalies in infected fetuses.Citation9, Citation10, Citation11 The rapidly expanding epidemic and this suspected congenital ZIKV syndrome have led the World Health Organization to declare the ZIKV outbreak as a global public health emergency on 1 February 2016.Citation12
The cause of the sudden emergence and rapid spread of ZIKV since 2007 is incompletely understood. A number of possible environmental factors have been proposed. First, globalization and urbanization have allowed ZIKV and its mosquito vectors to spread beyond their original geographical habitats. Second, major sport events including the World Cup and the Va’a World Sprint Championship canoe race in Brazil in 2014 might have provided an opportunity for infected travelers to introduce the virus to Latin America.Citation13 Third, climate changes associated with El Niño in South America in 2015 on the background trend of global warming possibly facilitated the rapid spread of Aedes mosquitoes and ZIKV.Citation14 Fourth, the increased awareness of and diagnostic capability for ZIKV infection likely led to the increased detection of this previously neglected disease. In contrast, little is known about the virological factors possibly associated with the apparent change in the spread of ZIKV after 2007. Although it has been shown that the epidemic strains are phylogenetically more closely related to the Asian than the African lineage of ZIKV, a comprehensive comparative analysis between the pre-epidemic and epidemic strains is lacking.Citation15 In this study, we performed comparative genomic analysis of all the pre-epidemic and epidemic strains with complete genome or complete polyprotein sequences available in GenBank to identify possible viral factors associated with this rapidly emerging viral epidemic.
MATERIALS AND METHODS
Viral sequences
The genome sequences of 24 ZIKV isolates with complete genome or complete polyprotein sequences available in GenBank (accessed on 18 February 2016) were included in this study (). These included strains collected from human, animals, and mosquitoes in Africa, Asia, the Pacific islands, and Latin America between 1947 and 2015. Representative genome sequences of other human-pathogenic flaviviruses, including Spondweni virus (SPOV, DQ859064.1), dengue virus serotype 2 (DENV-2, NC_001474.2), Japanese encephalitis virus (JEV, NC_001437.1), West Nile virus (WNV, NC_001563.2), yellow fever virus (YFV, NC_002031.1) and tick-borne encephalitis virus (TBEV, NC_001672.1) were also included.
Table 1 Genome sequences of Zika virus strains in this study
Genomic characterization and phylogenetic analysis
Phylogenetic tree construction by the maximum likelihood method was performed using MEGA 6.0 software, with bootstrap values being calculated from 500 trees. Protein family analysis was performed using the PFAM tool (http://pfam.xfam.org) Search for Conserved Domains server (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Prediction of transmembrane domains was performed using the TMHMM 2.0 server (http://www.cbs.dtu.dk/services/TMHMM/). Prediction of signal peptides was performed by using signalP software 4.1 (http://www.cbs.dtu.dk/services/SignalP/). The prediction of potential O-glycosylation and N-glycosylation sites in the polyprotein was performed using NetOGlyc 4.0 (http://www.cbs.dtu.dk/services/NetOGlyc/) and NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/), respectively. Secondary structure prediction in the 5′-untranslated region (UTR) was performed using the RNAfold WebServer with default settings (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). The number of synonymous substitutions per synonymous site, Ks, and the number of nonsynonymous substitutions per nonsynonymous site, Ka, for each coding region was calculated using the Nei and Gojobori substitution model with Jukes-Cantor correction in MEGA 6.0.Citation16, Citation17 Bootscan analysis was performed using Simplot version 3.5.1 as described previously,Citation18 with the Asian lineage of ZIKV strains as the query. Multiple alignment of the amino acid sequence of each protein are performed by ClustalX version 1.83 software and manually examined for any significant changes.
RESULTS
Genome arrangement
The single-stranded RNA genomes of the ZIKV strains used in this study range from 10 675 to 10 808 nucleotides encoding 3423 amino acids. The G+C content is 50.94%–51.26%. Similar to other flaviviruses, the ZIKV genomes have two flanking UTRs (5′- and 3′-UTR) and a single long open reading frame encoding a polyprotein. A type I cap structure without internal ribosomal entry site is present at the 5′ end followed by the conserved dinucleotide 5′-AG-3′. The 3′-end of the genome lacks a polyadenylate tail and terminates in a conserved 5′-CU-3′. The 5′- and 3′-UTRs are 107 and 429 nucleotides long, respectively. The 5′- and 3′-UTR sequences are conserved among ZIKV strains with nucleotide identities of ⩾83.6% and ⩾83.9%, respectively, but different from other flaviviruses including DENV, JEV, WNV, YFV, and TBEV (5′-UTR, ⩽69.5%; 3′-UTR, ⩽63.4%).
The polyprotein is cleaved into ten structural and non-structural (NS) proteins. Similar to other flaviviruses, the coding region orders and NS protein motifs of ZIKV are arranged in the order of 5′-Capsid (C)-preMembrane (prM)-Envelope (E)-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5-3′. Cleavage at the N terminus of the signal sequence for NS4B generates a 23 amino acid peptide (the 2 K peptides at amino acid position 2243–2265). The complete polyprotein sequences of ZIKV have low similarity with those of other human-pathogenic flaviviruses (DENV-2, 58.1% to 58.9%; SPOV, 68.3% to 69.0% nucleotide similarity). The ten structural and NS proteins contain multiple transmembrane domains that determine their location on the cytoplasmic or luminal side of the endoplasmic reticulum after cleavage (Figure 1). Most of the transmembrane domains are found in the NS2A, NS2B, NS4A, and NS4B proteins. This is compatible with the finding that the NS2A, NS2B, NS4A and NS4B proteins of other flaviviruses are mostly located within the endoplasmic reticular membrane bilayer, except for short regions between the transmembrane domains.Citation19 The other NS proteins include NS1, a putative protease/helicase (NS3), and a putative RNA-dependent RNA polymerase (RdRp; NS5), which are essential components or enzymes involved in viral replication.Citation19 Putative nuclear localization signals are found in C, NS1 and NS5 of both the pre-epidemic and epidemic strains, and NS3 of the prototype Uganda ZIKV strain (Figure 2). The putative O-glycosylation and N-glycosylation sites are mostly conserved, except for some variations at C, prM, E, NS1 and NS5 for O-glycosylation sites, and at E, NS2A and NS5 for N-glycosylation sites, which may be due to previous intracranial passage of the pre-epidemic Uganda and Malaysia ZIKV strains in mice.Citation20
Amino acid substitutions
Using all 24 available ZIKV genome sequences for analysis, the Ka/Ks ratios for the various coding regions were calculated (). Overall, the Ka/Ks ratios in ZIKV genomes are low, with the highest being observed at C coding region (0.077), suggesting that all the genes in the ZIKV genome are likely under stabilizing selection. Comparison between the pre-epidemic Asian lineage (Malaysia, 1966) and the epidemic ZIKV strains detected 24 amino acid substitutions (prM: 2, E: 3, NS2A: 1, NS3: 3, NS4B: 7 and NS5: 8) in the genomes of the latter virus strains (Figure 2). Four of these are associated with a change in the hydrophilicity/hydrophobicity of the amino acids (T773M in E, Y2082H in NS3, L2451S in NS4B and T2630V in NS5). Comparison between the pre-epidemic African lineage virus strains and the epidemic ZIKV strains detected 75 amino acid substitutions (C: 5, prM: 9, E: 10, NS1: 4, NS2A: 5, NS2B: 2, NS3: 9, NS4A: 1, NS4B: 9 and NS5: 21). Most of these are markers that differentiate the African and Asian lineages of ZIKV, which are also found in the pre-epidemic Asian lineage (Malaysia 1966) virus strain.Citation20 Fifteen substitutions are only present in the epidemic ZIKV strains and not the pre-2007 strains (V153M in prM; D679E, V759M, and T773M in E; A1285V in NS2A; M1970L in NS3; L2314F, V2449I and L2451S in NS4B; A2783V, N2892S, K3046R, P3158S, S3219D, and D3383N in NS5).
Phylogenetic relationship among all ZIKV strains
Phylogenetic analysis of the ten putative structural and NS coding regions showed that the ZIKV strains were clustered into the African and the Asian lineages (Figure 3). The epidemic ZIKV strains collected from the Pacific islands and South America clustered together with the Asian lineage strains. The complete polyprotein sequences of ZIKV within the same lineage (89.4%–99.8% nucleotide similarity) are more similar than those of different lineages (87.9%–95.5% nucleotide similarity). Notably, there is a change in the tree topology at the NS2B coding region, with a possible recombination occurring between ZIKV and SPOV.
Recombination analysis
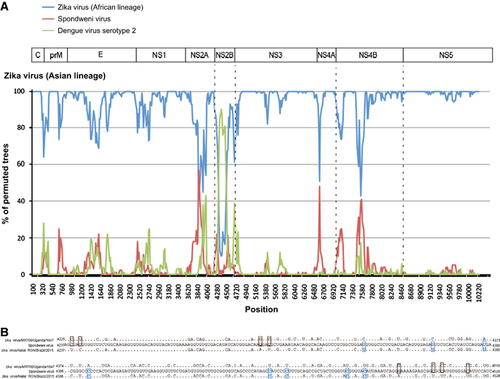
In view of the change in tree topology at the NS2B coding region, we performed recombination analysis to look for potential recombination sites in the ZIKV strains. From the 5′-end to the 3′-end of the genome, bootscan analysis showed a possible recombination fragment from nucleotide positions 4237–4528 between the Asian lineage ZIKV strains and SPOV, when the genomes of Asian lineage of ZIKV strains were used as the query and the African lineage of ZIKV strains, SPOV and DENV-2 were used as the reference (Figures 4A and 4B). This finding correlates with the change in tree topology at the NS2B gene in the phylogenetic tree.
Figure 1 Putative transmembrane domains in the Zika virus genome. Abbreviations: capsid, C; envelope, E; nonstructural, NS; pre-Membrane, prM.
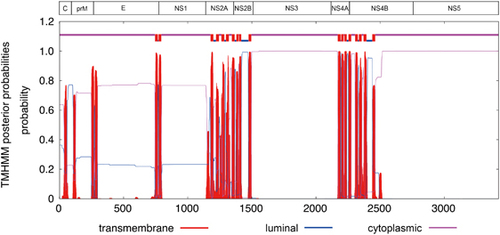
Figure 2 Comparative genomic analysis of the representative pre-epidemic and epidemic Zika virus strains. The results represent all 24 Zika virus strains unless otherwise specified (i.e., nuclear localization signals, and O-glycosylation and N-glycosylations sites). For the polymorphic sites, analysis was performed for all 24 Zika virus strains, but only the results of the three representative pre-epidemic African (Uganda 1947), pre-epidemic Asian (Malaysia 1966) and the epidemic (Brazil, 2015) strains are shown here. (A) Capsid (C), pre-Membrane (prM) and envelope (E); (B) nonstructural (NS) 1, NS2A, NS2B and NS3; (C) NS4A, NS4B and NS5. Abbreviation: DEAD-like helicase superfamily, DEXDc.

Table 2 Estimation of nonsynonymous and synonymous substitution rates in the 24 ZIKV genome sequences
RNA secondary structures and cyclization elements
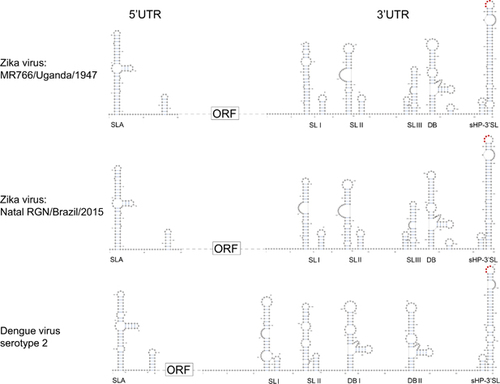
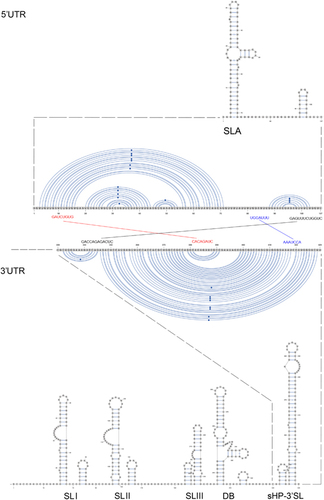
A Y-shape stem-loop A (SLA) structure is found at the 5′-end of the ZIKV genome (Figure 5). At the 3′-end of the viral genome, a small hairpin 3′-stem-loop (sHP-3′ SL) structure, three additional SL structures, and a dumbbell (DB) structure are found. Notably, the external loop of the SLI in domain 1 of the 3′-UTR just distal to the stop codon of the NS5 in the 1947 prototype pre-epidemic strain is replaced by a large bulge of nine nucleotide bases (UAG UCA GCC) in the representative epidemic ZIKV strain. Short conserved sequences within the 3′ terminal SL structure include the terminal 5′-CU-3′ and a 5′-ACAG-3′ in the top loop of the sHP-3′ SL structure. There are three pairs of inverted complementary sequences (GAU CUG UG-CAC AGA UC, UGG AUU U-AAA UCC A and GAG UUU CUG GUC-GAC CAG AGA CUC and GAG UUU CUG GUC-GAC CAG AGA CUC that may mediate genome cyclization (Figure 6).
Figure 3 Phylogenetic analysis of the ten structural and non-structural coding regions of 24 Zika virus strains, rooted by Spondweni virus, dengue virus serotype 2, and tick-borne encephalitis virus. The trees were constructed by the maximum likelihood method based on the Tamura-Nei model. The tree with the highest log likelihood is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Joining and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood approach, and then selecting the topology with superior log likelihood value. The bootstrap values were calculated from 500 trees. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The bootstrap values <60% are not shown. All Zika virus strains are labeled as follow: accession number/strain number/country/year. All strains represent Zika virus strains unless otherwise specified for Spondweni virus, dengue virus serotype 2 and tick-borne encephalitis virus. Abbreviation: nonstructural, NS.
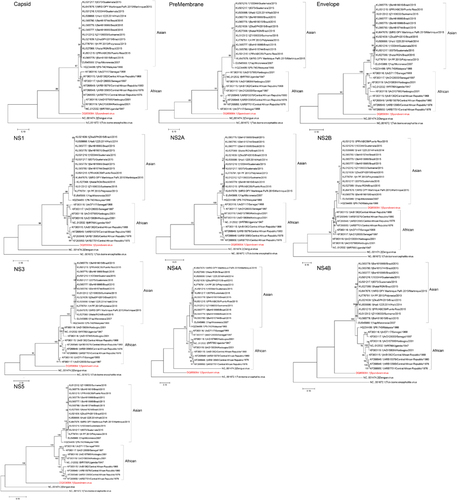
Figure 4 Genome organization and bootscan analysis of the Zika virus genomes. (A) Bootscanning was conducted with Simplot version 3.5.1 on a gapless nucleotide alignment, which was generated with ClustalX with the genome sequences of the available Asian lineage Zika virus strains as the query sequences. (B) Multiple alignment of the recombination fragment in NS2B nucleotide sequences of Zika virus strains MR766/Uganda/1947 (NC_012532.1) and Natal RGN/Brazil/2015 (KU527068.1), and Spondweni virus (DQ859064.1). In the Zika virus strains, only the nucleotides differing from those in Spondweni virus are depicted. The nucleotides in the Zika virus strains are highlighted in blue or red. Abbreviations: capsid, C; envelope, E; nonstructural, NS; pre-Membrane, prM.
DISCUSSION
As demonstrated in recent epidemics of emerging viral infections, characterization of the viral genome may facilitate the identification of important virulence factors and diagnostic, therapeutic and vaccine targets.Citation21, Citation22, Citation23 In this study, we analyzed the available genomic data of ZIKV in GenBank to provide a quick search for possible virus mutations that may be associated with the rapidly expanding ZIKV epidemic.
Our genomic analysis revealed some changes in the 3′-UTR sequence of the post-2007 epidemic ZIKV strain. The 5′ and 3′ terminal sequences of the genome of flaviviruses fold into conserved RNA secondary structures and encode regions essential for genome cyclization at the initial phase of replication.Citation24 In mosquito-borne flaviviruses including ZIKV, the 3′ UTR is further divided into three domains, including the highly variable proximal domain 1 that directly follows the stop codon, the moderately conserved domain 2 that contains the SL and DB structures, and the highly conserved domain 3 that contains the complementary cyclization elements and the conserved sHP-3′ SL structure. Deletion of the SL sequences in the 5′- or 3′-UTR is lethal for flavivirus infectious clones.Citation25, Citation26 These secondary RNA structures bind to host proteins, such as elongation factor 1α and poly(A)-binding protein, and proteins of the viral replication complex, including C, NS2A, NS3 and NS5 proteins, to promote genomic RNA cyclization.Citation27 Genomic RNA cyclization is essential for viral replication in two ways. First, the 5′-SLA acts as a promotor element to stimulate the NS5 RdRp to initiate negative strand synthesis at the 3′-UTR.Citation28, Citation29, Citation30 Second, the 5′- and 3′-UTRs move into close proximity for cap-dependent translation of the viral polyprotein to proceed.Citation31, Citation32, Citation33
The two most conserved secondary RNA structures in flavivirus genomes are the Y-shape SLA structure at the 5′-UTR and the sHP-3′ SL structure at the 3′-UTR.Citation24 Expectedly, these are also present in the ZIKV genomes. The arrangement and sequences of the other 3′-UTR RNA secondary structures of ZIKV are less conserved from those of other flaviviruses.Citation24 For example, there are two DB structures and two SL structures in addition to the conserved sHP-3′ SL structure in DENV-1 and DENV-3, whereas three additional putative SL and one DB structures are found in both the pre-epidemic and epidemic ZIKV strains.Citation24 Interestingly, we found a large bulge of nine nucleotide bases at the SLI of the epidemic ZIKV strain, which more closely resembles the SLII than the corresponding SLI of the pre-epidemic strain. Wet laboratory experiments are required to investigate the functional role of this putative conformational change in the transmissibility and virulence of the epidemic ZIKV strain.
Figure 5 Schematic representations of the Zika virus genome RNA secondary structures. The short conserved 5′-ACAG-3′ sequences in the top loop of the sHP-3′ SL structure are indicated in red. Abbreviations: dumbbell, DB; open reading frame, sHP-3'-ORF; small hairpin 3'-stem-loop, sHP-3' SL; stem loop, SL; Y-shape stem-loop, SLA; untranslated region, UTR.
After cleavage of the polyprotein, the C protein of flaviviruses is released into the cytoplasm and forms homodimers. The basic residues on one side of the C protein bind the RNA genome and the hydrophobic residues on the other side interact with the viral lipid envelope.Citation34, Citation35 Even after virus-endosomal membrane fusion, the entering viral genome may remain associated with the C dimers to evade from host nucleases and RNA sensors. Thus, the C protein of flaviviruses may function as an RNA chaperone in addition to its role in the formation of viral nucleocapsid. The resulting nucleocapsid then buds into the endoplasmic reticular lumen to form viral particles with the prM and E proteins.Citation36, Citation37 The C protein may also be found in the nuclei and nucleoli of cell lines infected by flaviviruses, including DENV-2, DENV-4, WNV, JEV and Kunjin virus.Citation38, Citation39, Citation40 The migration of the C protein to the nuclei and nucleoli are believed to be mediated by nuclear localization signals.Citation41 In ZIKV, we found one putative nuclear localization signal near the 3′-end of the C protein, which is conserved in the pre-epidemic and epidemic strains (Figure 2A). JEV with a single point mutation (T45G) at the N terminus of the C coding region has reduced virulence.Citation42 In our study, we found five amino acid substitutions in C protein including N25S, L27F, R101K, I110V and I113V, which were detected in the Asian ZIKV strains as compared with the prototype Uganda African ZIKV strain. The importance of these findings should be verified in future experiments.
The prM protein of flaviviruses interacts with the E protein to form prM-E heterodimers, which are essential for the formation of immature virions. The prM protein is then cleaved to M protein through cellular proteases to produce and release mature virions. The M protein of flaviviruses contains two membrane-spanning domains and a short ectodomain. Histidine at residue 99 and the transmembrane region of the prM protein of JEV is critical for stable prM-E heterodimeric complex formation.Citation43 Moreover, a single amino acid substitution at the N-linked glycosylation site of the prM-E complex of JEV may elicit an enhanced host humoral immune response, which could be a useful strategy for vaccine design.Citation44 The functional roles of the V153M substitution, which is found in all the ZIKV epidemic strains, and other amino acid substitutions I125V, S139N, K143E, A148P, H157Y, V158I, K246R and V262A, which are different between the African and Asian lineages, may be of some importance in future studies.
The E protein is the major surface protein of flaviviruses and is involved in viral attachment, fusion, penetration, hemagglutination, host range and cell tropism.Citation45 Structurally, three domains could be found in the E protein of flaviviruses based on X-ray crystallographic structural studies. Domain I is located in the middle of the E protein and contains the N terminus with glycosylation sites. Domain II contains the fusion peptide at the distal side and flanks one side of domain I. Domain III flanks the other side of domain I and is the major antigenic region in the E protein. It also contains the receptor-binding site and is, therefore, an important therapeutic and vaccine target of flaviviruses. E345K substitution at the E protein of DENV-4 is associated with reduced viral virulence.Citation46 Importantly, we found a number of amino acid substitutions in the E protein of the epidemic ZIKV strain (Figure 2A). Among these substitutions, V603I and D679E are found in the domain III of the E protein. The I (isoleucine) at position 603 and the E (glutamic acid) at position 679 are present in all of the epidemic strains, but in none of the pre-epidemic strains. Investigation on the presence and function of these amino acid substitutions should be performed on a larger collection of epidemic strains.
Figure 6 Terminal region genome sequences that are involved in 3′–5′ long distance RNA–RNA interactions. The three pairs of inverted complementary sequences that may mediate genome cyclization and allow the RdRp to reach the 3′ SL initiation site for RNA synthesis are enlarged. Abbreviations: dumbbell, DB; open reading frame, sHP-3' ORF; small hairpin 3'-stem-loop, sHP-3'-SL; stem Loop, SL; Y-shape stem-loop, SLA; untranslated region, UTR.
NS1 of flaviviruses is a glycoprotein that may contain multiple N-glycosylation sites and disulfide bonds that may affect virus viability and virulence.Citation47 Significant NS1 codon usage adaptation to human housekeeping genes by the recent Asian lineage of ZIKV has been suggested to be a facilitator of viral replication and increased viral titers.Citation48 NS1 may exist in different forms, depending on the variable formation of N-glycosylation sites and disulfide bonds. The monomers of NS1 are soluble and hydrophilic, whereas the NS1 homodimers may associate with the endoplasmic reticular membranes.Citation49, Citation50 The NS1 protein also exists in a soluble hexamer form that is secreted by mammalian cells.Citation19 Mutations at the NS1 N-glycosylation sites may significantly affect viral replication and virulence in YFV.Citation51 Deletion studies suggest that NS1 is required for initiation of RNA synthesis and especially during early negative strand RNA genome synthesis.Citation19 Using the available genomic data in this study, we were not able to find any putative N-glycosylation sites in NS1 of the ZIKV genomes, but we found four different amino acid substitutions between the African and the Asian lineages (E842D, K859R, A984V and V1026I). The importance of these substitutions will await further verifications.
NS2A, NS2B, NS4A and NS4B of flaviviruses are small, hydrophobic proteins with incompletely understood functions and no known enzymatic motifs.Citation19 Each of these proteins has two or more membrane-spanning regions and may play important roles in the assembly or anchoring of the viral replication complexes on the endoplasmic reticular membrane.Citation52, Citation53 Mutation at the helix-breaker amino acid R84 at the NS2A of WNV may attenuate viral replication.Citation19 NS2B interacts with the NS3 C-terminal protease domain to form the viral serine protease complex that is involved in the cleavage of the viral polyprotein. Moreover, these NS proteins may exert important effects on the host immune response. The DENV NS2B/NS3 complex has been shown to mediate cleavage of STING, which is a key mediator in the pathways of the host innate immune response.Citation54 Expression of WNV and Kunjin virus NS2A, NS2B, NS4A and NS4B proteins may also block type I interferon signaling.Citation55, Citation56 Mutations at the NS4B of WNV may attenuate the neurovirulence and viral replication in infected mice.Citation57, Citation58 We found two amino acid substitutions that are present in all the epidemic strains, including V2449I and L2451S, among the analyzed ZIKV strains (Figure 2C). Interestingly, we also found that there is likely a recombination between the NS2B of ZIKV and SPOV, although the direction of gene transfer was uncertain (Figures 3 and 4). A recent study did not detect recombination events between the epidemic Brazilian ZIKV strain and other arboviruses including DENV-1, DENV-4, WNV, YFV and Chikungunya virus, but SPOV was not included in the analysis.Citation10 Further investigations are necessary to determine the biological significance of these amino acid substitutions among the ZIKV strains and this recombination between the two human-pathogenic mosquito-borne flaviviruses.
The major viral enzymes of flaviviruses are encoded by the NS3 and NS5 coding regions. In addition to its putative protease activity at the N terminus, the C-terminal domain of the NS3 protein of ZIKV also possesses putative ATPase/helicase, nucleoside triphosphatase, and 5′-triphosphatase activities. The multiple roles of NS3 in the viral replication cycle make the protein an attractive antiviral target. NS5 is the largest and most conserved protein of ZIKV. The N-terminal part contains a putative methyltransferase domain with both N7 and 2′-O-methyltransferase and guanyltransferase activities. The C-terminal part contains typical motifs of RdRp as in other flaviviruses. The NS5 protein of DENV mediates cleavage of STAT2, which is an important mediator in the host innate immune response signaling pathways.Citation59 In this study, we found that NS5, being the largest protein, also has the largest number of amino acid substitutions (n=21) when the strains of African and Asian lineages are compared. Eight amino acid substitutions in the NS3 and NS5 are different between the pre-epidemic and epidemic strains, including M1970L, T2630V, A2783V, N2892S, K3046R, P3158S, S3219D and D3383N. The importance of these substitutions should be investigated further.
Although ZIKV was estimated to have emerged between 1892 and 1943 by Bayesian evolutionary analysis and has been isolated for nearly 70 years, very little is known about the virology of this emerging virus.Citation60 In this study, we summarized its genomic changes with a limited number of virus strains. These virus strains included both the pre-epidemic African and Asian lineage strains that were mostly mosquito isolates, and the epidemic strains found in human. Among these virus strains, we have detected a number of amino acid substitutions throughout the genome and a conformational change in the SLI structure at the 3′-UTR of the epidemic ZIKV strain. We have also detected a possible recombination of a NS2B fragment between the Asian lineage of ZIKV and SPOV. The impact of these changes of the virus genome on the virulence, viability and transmissibility of ZIKV should be further investigated in biological assays.
We thank James Cai (Department of Veterinary Integrative Biosciences, Texas A&M University) and Yu-Mei Wen (Key Laboratory of Medical Molecular Virology, Fudan University) for facilitation of the study. The study was partly supported by the Consultancy Service for Enhancing Laboratory Surveillance of Emerging Infectious Disease of the Department of Health, the Food and Health Bureau, Hong Kong Special Administrative Region, China; the Key Laboratory of Medical Molecular Virology, Fudan University; the Croucher Senior Medical Research Fellowship; and the Strategic Research Theme Fund, The University of Hong Kong; and by the donations of Mr Larry Chi-Kin Yung and Hui Hoy & Chow Sin Lan Charity Fund Limited. The funding sources had no role in study design; data collection, analysis or interpretation; or writing of the report. Jasper Fuk-Woo Chan and Kwok-Yung Yuen had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Related Research Data
- ChanJF,ChoiGK,YipCCet al.Zika fever and congenital Zika syndrome: an unexpected emerging arboviral disease?J Infect2016e-pub ahead of print 2 March 2016; doi:https://doi.org/10.1016/j.jinf.2016.02.011..
- DickGW,KitchenSF,HaddowAJ.Zika virus. I. Isolations and serological specificity.Trans R Soc Trop Med Hyg1952; 46:509–520.
- DuffyMR,ChenTH,HancockWTet al.Zika virus outbreak on Yap Island, Federated States of Micronesia.N Engl J Med2009; 360:2536–2543.
- TognarelliJ,UlloaS,VillagraEet al.A report on the outbreak of Zika virus on Easter Island, South Pacific, 2014.Arch Virol2016; 151:665–668.
- RothA,MercierA,LepersCet al.Concurrent outbreaks of dengue, chikungunya and Zika virus infections - an unprecedented epidemic wave of mosquito-borne viruses in the Pacific 2012-2014.Euro Surveill2014; 19:pii: 20929.
- Dupont-RouzeyrolM,O’ConnorO,CalvezEet al.Co-infection with Zika and dengue viruses in 2 patients, New Caledonia, 2014.Emerg Infect Dis2015; 21:381–382.
- BogochII,BradyOJ,KraemerMUet al.Anticipating the international spread of Zika virus from Brazil.Lancet2016; 387:335–336.
- European Centre for Disease Prevention and Control (ECDC).Rapid Risk Assessment - Zika virus epidemic in the Americas: potential association with microcephaly and Guillain-Barré syndrome. Solna: ECDC, 2015. Available athttp://ecdc.europa.eu/en/publications/Publications/zika-virus-americas-association-with-microcephaly-rapid-risk-assessment.pdf(accessed on 20 February 2016).
- MlakarJ,KorvaM,TulNet al.Zika Virus Associated with Microcephaly.N Engl J Med2016e-pub ahead of print 10 February 2016; doi:https://doi.org/10.1056/NEJMoa1600651.
- CalvetG,AguiarRS,MeloASet al.Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: a case study.Lancet Infect Dis2016e-pub ahead of print 17 February 2016; doi:https://doi.org/10.1016/S1473-3099(16)00095-5.
- Schuler-FacciniL,RibeiroEM,FeitosaIMet al.Possible Association Between Zika Virus Infection and Microcephaly - Brazil, 2015.Morbid Mortal Wkly Rep2016; 65:59–62.
- World Health Organization (WHO)WHO statement on the first meeting of the International Health Regulations (2005) (IHR 2005) Emergency Committee on Zika virus and observed increase in neurological disorders and neonatal malformations; 1 February 2016.Geneva: WHO, 2016. Available athttp://www.who.int/mediacentre/news/statements/2016/1st-emergency-committee-zika/en/(accessed on 20 February 2016).
- SalvadorFS,FujitaDM.Entry routes for Zika virus in Brazil after 2014 world cup: New possibilities.Travel Med Infect Dis2016; 14:49–51.
- PazS,SemenzaJC.El Niño and climate change-contributing factors in the dispersal of Zika virus in the Americas?Lancet2016; 387:745.
- EnfissiA,CodringtonJ,RoosbladJet al.Zika virus genome from the Americas.Lancet2016; 387:227–228.
- NeiM,GojoboriT.Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions.Mol Biol Evol1986; 3:418–426.
- TamuraK,StecherG,PetersonDet al.MEGA6: Molecular Evolutionary Genetics Analysis version 6.0.Mol Biol Evol2013; 30:2725–2729.
- LauSK,LeeP,TsangAKet al.Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination.J Virol2011; 85:11325–1137.
- BrintonMA.Replication cycle and molecular biology of the West Nile virus.Viruses2014; 6:13–53.
- HaddowAD,SchuhAJ,YasudaCYet al.Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage.PLoS Negl Trop Dis2012; 6:e1477.
- ChanJF,LauSK,ToKKet al.Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease.Clinical Microbiol Rev2015; 28:465–522.
- ChanJF,ToKK,TseHet al.Interspecies transmission and emergence of novel viruses: lessons from bats and birds.Trends Microbiol2013; 21:544–555.
- ZumlaA,ChanJF,AzharEIet al.Coronaviruses—drug discovery and therapeutic options.Nat Rev Drug Discov2016e-pub ahead of pinrt 12 February 2016; doi:https://doi.org/10.1038/nrd.2015.37.
- VillordoSM,CarballedaJM,FilomatoriCVet al.RNA Structure Duplications and Flavivirus Host Adaptation.Trends Microbiol2016e-pub ahead of pinrt 2 February 2016; doi:https://doi.org/10.1016/j.tim.2016.01.002.
- CahourA,PletnevA,Vazielle-FalcozMet al.Growth-restricted dengue virus mutants containing deletions in the 5′ noncoding region of the RNA genome.Virology1995; 207:68–76.
- YuL,MarkoffL.The topology of bulges in the long stem of the flavivirus 3′ stem-loop is a major determinant of RNA replication competence.J Virol2005; 79:2309–2324.
- RobyJA,PijlmanGP,WiluszJet al.Noncoding subgenomic flavivirus RNA: multiple functions in West Nile virus pathogenesis and modulation of host responses.Viruses2014; 6:404–427.
- FilomatoriCV,LodeiroMF,AlvarezDEet al.A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome.Genes Dev2006; 20:2238–2249.
- DongH,ZhangB,ShiPY.Flavivirus methyltransferase: a novel antiviral target.Antiviral Res2008; 80:1–10.
- ZhangB,DongH,SteinDAet al.West Nile virus genome cyclization and RNA replication require two pairs of long-distance RNA interactions.Virology2008; 373:1–13.
- PolacekC,FriebeP,HarrisE.Poly(A)-binding protein binds to the non-polyadenylated 3′ untranslated region of dengue virus and modulates translation efficiency.J Gen Virol2009; 90:687–692.
- BlackwellJL,BrintonMA.Translation elongation factor-1 alpha interacts with the 3′ stem-loop region of West Nile virus genomic RNA.J Virol1997; 71:6433–6444.
- De Nova-OcampoM,Villegas-SepulvedaN,del AngelRM.Translation elongation factor-1alpha, La, and PTB interact with the 3′ untranslated region of dengue 4 virus RNA.Virology2002; 295:337–347.
- LobigsM.Flavivirus premembrane protein cleavage and spike heterodimer secretion require the function of the viral proteinase NS3.Proc Natl Acad Sci USA1993; 90:6218–6222.
- AmbergSM,NestorowiczA,McCourtDWet al.NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: in vitro and in vivo studies.J Virol1994; 68:3794–3802.
- MaL,JonesCT,GroeschTDet al.Solution structure of dengue virus capsid protein reveals another fold.Proc Natl Acad Sci USA2004; 101:3414–3419.
- WangSH,SyuWJ,HuST.Identification of the homotypic interaction domain of the core protein of dengue virus type 2.J Gen Virol2004; 85:2307–2314.
- WestawayEG,KhromykhAA,KenneyMTet al.Proteins C and NS4B of the flavivirus Kunjin translocate independently into the nucleus.Virology1997; 234:31–41.
- MoriY,OkabayashiT,YamashitaTet al.Nuclear localization of Japanese encephalitis virus core protein enhances viral replication.J Virol2005; 79:3448–3458.
- OhW,YangMR,LeeEWet al.Jab1 mediates cytoplasmic localization and degradation of West Nile virus capsid protein.J Biol Chem2006; 281:30166–30174.
- SangiambutS,KeelapangP,AaskovJet al.Multiple regions in dengue virus capsid protein contribute to nuclear localization during virus infection.J Gen Virol2008; 89:1254–1264.
- JiaF,ZhuX,XuF.A single adaptive point mutation in Japanese encephalitis virus capsid is sufficient to render the virus as a stable vector for gene delivery.Virology2016; 490:109–118.
- LinYJ,WuSC.Histidine at residue 99 and the transmembrane region of the precursor membrane prM protein are important for the prM-E heterodimeric complex formation of Japanese encephalitis virus.J Virol2005; 79:8535–8544.
- ZhangY,ChenP,CaoRet al.Mutation of putative N-linked glycosylation sites in Japanese encephalitis virus premembrane and envelope proteins enhances humoral immunity in BALB/C mice after DNA vaccination.Virol J2011; 8:138.
- ChavezJH,SilvaJR,AmarillaAAet al.Domain III peptides from flavivirus envelope protein are useful antigens for serologic diagnosis and targets for immunization.Biologicals2010; 38:613–618.
- LinHH,LeeHC,LiXFet al.Dengue type four viruses with E-Glu345Lys adaptive mutation from MRC-5 cells induce low viremia but elicit potent neutralizing antibodies in rhesus monkeys.PLoS One2014; 9:e100130.
- WhitemanMC,WickerJA,KinneyRMet al.Multiple amino acid changes at the first glycosylation motif in NS1 protein of West Nile virus are necessary for complete attenuation for mouse neuroinvasiveness.Vaccine2011; 29:9702–9710.
- de Melo FreireCC,IamarinoA,de Lima NetoDFet al.Spread of the pandemic Zika virus lineage is associated with NS1 codon usage adaptation in humans.bioRxiv2015e-pub ahead of print 25 November 2015; doi:http://dx.doi.org/10.1101/032839.
- WinklerG,MaxwellSE,RuemmlerCet al.Newly synthesized dengue-2 virus nonstructural protein NS1 is a soluble protein but becomes partially hydrophobic and membrane-associated after dimerization.Virology1989; 171:302–305.
- WinklerG,RandolphVB,CleavesGRet al.Evidence that the mature form of the flavivirus nonstructural protein NS1 is a dimer.Virology1988; 162:187–196.
- MuylaertIR,GallerR,RiceCM.Genetic analysis of the yellow fever virus NS1 protein: identification of a temperature-sensitive mutation which blocks RNA accumulation.J Virol1997; 71:291–298.
- MackenzieJM,KhromykhAA,JonesMKet al.Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A.Virology1998; 245:203–215.
- MillerS,SparacioS,BartenschlagerR.Subcellular localization and membrane topology of the Dengue virus type 2 Non-structural protein 4B.J Biol Chem2006; 281:8854–8863.
- AguirreS,MaestreAM,PagniSet al.DENV inhibits type I IFN production in infected cells by cleaving human STING.PLoS Pathog2012; 8:e1002934.
- LiuWJ,WangXJ,MokhonovVVet al.Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins.J Virol2005; 79:1934–1942.
- LiuWJ,WangXJ,ClarkDCet al.A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice.J Virol2006; 80:2396–2404.
- WickerJA,WhitemanMC,BeasleyDWet al.A single amino acid substitution in the central portion of the West Nile virus NS4B protein confers a highly attenuated phenotype in mice.Virology2006; 349:245–253.
- WickerJA,WhitemanMC,BeasleyDWet al.Mutational analysis of the West Nile virus NS4B protein.Virology2012; 426:22–33.
- AshourJ,Laurent-RolleM,ShiPYet al.NS5 of dengue virus mediates STAT2 binding and degradation.J Virol2009; 83:5408–5418.
- FayeO,FreireCC,IamarinoAet al.Molecular evolution of Zika virus during its emergence in the 20(th) century.PLoS Negl Trop Dis2014; 8:e2636.