
Abstract
Emerging Microbes & Infections (2017) 6, e54; doi:10.1038/emi.2017.39; published online 21 June 2017
Dear Editor,
Viral encephalitis (VE) and viral meningitis (VM) are severe infectious diseases that cause substantial morbidity and mortality in children. Non-polio human enteroviruses (EVs), especially the species Enterovirus B (EV-B), are major causative agents of these diseases. Echovirus 18 (E18), a serotype of EV-B, has mostly been reported as an important pathogen that has caused many outbreaks of aseptic meningitis in the United States,Citation1, Citation2 Japan,Citation3 Korea,Citation4 Germany,Citation5 and other countries or regions.Citation6 So far, there has been no report of an E18-associated encephalitis/meningitis outbreak in mainland China, but little E18 epidemiological data is available.Citation7 Here we describe an outbreak of E18 encephalitis/meningitis in children that occurred in Hebei Province, China, in 2015.
From April 2015, a marked increase in the number of children with VE/VM was observed in Hebei Province, in the central part of China, whereas only three cases were found from January to March 2015. Therefore, we conducted a study to explore the viral pathogens in these encephalitis/meningitis cases. A total of 268 pediatric VE/VM cases (190 male and 78 female) were enrolled in this study from January to September. The median age of the patients was 5.81 years, with the range being 7 months to 16 years (mean age, 6.11±2.70 years). Most patients (83.6%, 224/268) were 3–10 years old, and no neonates were enrolled in this study. The most predominant clinical manifestations were headache (89.9%, 241/268), vomiting (81.3%, 218/268), fever (62.7%, 168/268), lethargy (26.9%, 72/268) and nausea (22.0%, 59/268), whereas some patients had abdominalgia (9.0%, 24/268), convulsions (3.7%, 10/268) and confusion (2.6%, 7/268). All patients recovered, except for one patient who had secondary epilepsy.
Cerebrospinal fluid (CSF) samples were collected from every case. Viral RNA was extracted from the CSF with a QIAamp MinElute Virus Spin Kit (Qiagen, Hilden, Germany). Viral pathogens, including EVs, Japanese encephalitis virus and mumps virus, were screened using real-time PCR. Herpes viruses, including herpes simplex virus 1 (HSV1), HSV2, varicella zoster virus, Epstein–Barr virus, human herpes virus 6 and cytomegalovirus, were screened by multiple PCR methods with the Seeplex meningitis-V1 ACE Detection kit (Seegene, Seoul, Korea). Partial VP1 genes from EVs were amplified with degenerate primers.Citation8, Citation9 EV genotyping was done by sequencing the partial VP1 genes and performing BLAST analysis. The complete VP1 gene sequences of E18 were obtained by further amplification and sequencing with E18 VP1-specific primers. Nucleotide sequence alignments and identities were implemented in MEGA5.03 software (version 5.0; Sudhir Kumar, Arizona State University, Tempe, AZ, USA).Citation10 Phylogenetic analysis was conducted on the complete VP1 gene sequences (this study and GenBank database) by using Bayesian Evolutionary Analysis Sampling Trees (BEAST) version 1.7 software.Citation11 Potential recombination of the E18 strain was scanned using a similarity plot and bootscan method with the Simplot program (version 3.5.1; Stuart Ray, Johns Hopkins University, Baltimore, MD, USA).Citation12 According to the results of the screening, the EV-positive samples were cultured on rhabdomyosarcoma cell lines for virus isolation. After high titer virus was obtained, a representative strain was selected to sequence the complete genome.
The total positive detection rate of viruses was 64.9% (174/268), EVs accounted for 51.1% (89/174), followed by HSV1 (19.5%, 34/174), varicella zoster virus (10.9%, 19/174), HSV2 (6.3%, 11/174), Epstein–Barr virus (4.6%, 8/174), cytomegalovirus (4.0%, 7/174) and human herpes virus 6 (3.4%, 6/174). No Japanese encephalitis virus and mumps virus-positive samples were detected. The cases peaked in July and August (Supplementary Figure S1). Among the 82 genotyped EVs, E18 was the most predominant, accounting for 74.4% (61/82), followed by E30 (17.1%, 14/82), E6 (3.7%, 3/82), E14 (1.2%, 1/82), E33 (1.2%), coxsackievirus A9 (CV-A9; 1.2%) and CV-B5 (1.2%). The genotypes of seven EVs were not confirmed because of poor sequence data quality. A total of 57 E18 complete VP1 gene sequences were obtained (accession number KY303773–KY303829). Seventy-one EV-positive CSF samples were cultured with rhabdomyosarcoma cell, and 29 EV strains were obtained, including 18 strains of E18, 8 strains of E30 and 3 strains of E6 (Supplementary Table S1). The remaining 42 CSF samples had negative cultures.
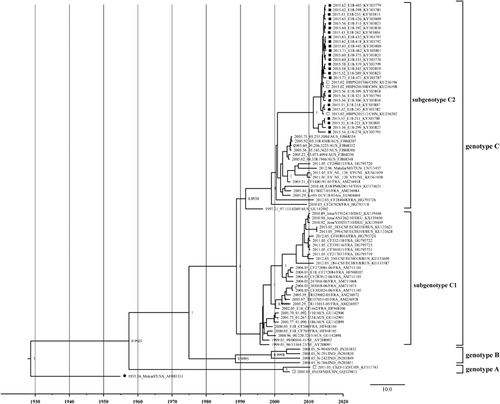
A phylogenetic tree based on the complete VP1 gene sequences was constructed by the BEAST software (). Referring to the genotyping criteria of CV-A16,Citation13 more than 15% of the nucleotide difference in the complete VP1 region of the E18 strain was used to distinguish the genotypes in this study. According to the results, except for the E18 prototype strain (Metcalf, accession number AF317694), all the E18 strains could be divided into three genotypes—A, B and C—with the support of the high confidence values (posterior value >99%). The mean genetic distance between genotypes A and B was 0.208, A and C was 0.245, and B and C was 0.218. The nucleotide identities were 80.1%–83% (A and B), 76.5%–81.4% (A and C) and 79.4%–84.3% (B and C). The prototype Metcalf shared 79.6%–79.7%, 78.9%–80.8% and 77.1%–80.3% nucleotide identity with genotypes A, B and C, respectively. The two strains of genotype A were in China in 2005 and 2011, respectively. Genotype B was collected in India in 2008, and the strains shared 95.8%–98.6% nucleotide identity. Genotype C can be further divided into two subgenotypes (C1 and C2), with the support of high confidence values (posterior value >99%) and a mean genetic distance of 0.138 between the subtypes. All isolates from Hebei Province in 2015 clustered into the C2 subtype. The strains in the C1 subtype shared 90%–100% nucleotide identity and 92.3%–100% amino-acid identity with each other. The strains in the C2 subtype shared 86.7%–100% nucleotide identity and 92.3%–100% amino-acid identity with each other.
Figure 1 The phylogenetic tree based on the alignments of the complete VP1 gene sequences of E18 was constructed with BEAST software (100 million generations) using a lognormal relaxed clock, a constant size tree prior and the GTR+G substitution model. Squares indicate China E18, whereas filled squares indicate the sequences from this study. Filled circle indicates the E18 Metcalf prototype strain. The scale bar indicates years. The strain name, year of sampling and GenBank accession numbers are presented.
The B–C loop, located from amino acid 78–89 in the E18 VP1 region, is important for the reactivity of type-specific neutralizing antibodies, which formed a part of the epitope on the surface of the virion.Citation14 There were 18 amino-acid substitutions between the prototype strain and the strains in this study. One amino-acid substitution was located at residue 84 (R→N) in the B-C loop. These amino-acid changes might be a significant cause of the prevalence of E18, but this will require intensive studies.
The complete genome sequence of the E18–314 isolate was available and was analyzed (accession number KX767786). The nucleotide identity of the complete genomes was 80.6% compared with the prototype strain, Metcalf. The nucleotide identities of the 5′UTR, P1, P2, P3, 3′UTR and VP1 regions of E18–314 compared with the prototype strain were 81.5%, 80.2%, 79.7%, 81.0%, 87.1% and 80.7%, respectively.Citation15 The EV-B prototype strains were used as reference sequences, and similarity plots and bootscanning analyses were performed to depict the possible recombination of the E18 strain. The results showed the highest similarity and nearest phylogenetic relationship with the E18 prototype in the P1 region. However, in the 5′UTR, P2 and P3 regions, the E18–314 strain contained some regions that showed higher similarity with the E9, CV-B4, EV-B106, E13 and E16 prototypes, but apparently not the Metcalf strain. Two obvious crossing sites in the 5′UTR and 2A regions suggested that a potential recombination occurred (Supplementary Figure S2).
These results indicated that there was an outbreak of E18 encephalitis/meningitis in children in Hebei Province, China, in 2015. The phylogenetic analysis suggests that the outbreak was caused by a new E18 strain. The representative strain, E18–314, was a potential multiple-recombinant virus, which contained other EV-B donor sequences. Genetic recombination is sometimes an important reason for changes in enterovirus virulence and can trigger serious public health problems.
Little data on the molecular epidemiology of E18 in mainland China has been obtained before the current study. Further study is needed to gain a better insight into the prevalence of E18 infection in mainland China.
Supplementary Figure S1
Download MS Word (47 KB)Supplementary Figure S2
Download MS Word (874.5 KB)Supplementary Table S1
Download MS Word (120.5 KB)Acknowledgments
This study was approved by the Medical Ethics Committee of the Beijing Children’s Hospital affiliated with Capital Medical University, and informed consent was obtained. We thank Dr Getu Zhaori (Honorary Editor-in-chief, Chinese Medical Journal, Chinese Medical Association, Beijing, China) for assistance in polishing the manuscript. We also thank all the reviewers for their comments, which improved this manuscript. This work was supported by the National Science and Technology Major Projects for the Control and Prevention of Major Infectious Diseases in China (Grant NO 2013ZX10004-202); the Beijing Outstanding Young Talent Training Projects (Grant NO 2014000021469G237); the Capital Clinical Feature Project of Beijing Technology Program (Grant NO Z151100004015046); and the Basic and Clinical Research Cooperation Project of Capital Medical University (Grant NO 17JL11). The sponsors had no role in the study design, data analysis, manuscript preparation or publishing decision.
Supplementary Information for this article can be found on the Emerging Microbes & Infections website (http://www.nature.com/emi)
Related Research Data
References
- Wilfert CM, Lauer BA, Cohen Met al.An epidemic of echovirus 18 meningitis. J Infect Dis 1975;131: 75–78.
- Turabelidze G, Lin M, Butler Cet al.Outbreak of echovirus 18 meningitis in a rural Missouri community. Mo Med 2009;106: 420–424.
- Miyamura K, Yamashita K, Yamadera Set al.An epidemic of echovirus 18 in 1988 in Japan—high association with clinical manifestation of exanthem. A report of the National Epidemiological Surveillance of Infectious Agents in Japan. Jpn J Med Sci Biol 1990;43: 51–58.
- Baek K, Park K, Jung Eet al.Molecular and epidemiological characterization of enteroviruses isolated in Chungnam, Korea from 2005 to 2006. J Microbiol Biotechnol 2009;19: 1055–1064.
- Krumbholz A, Egerer R, Braun Het al.Analysis of an echovirus 18 outbreak in Thuringia, Germany: insights into the molecular epidemiology and evolution of several enterovirus species B members. Med Microbiol Immunol 2016;205: 471–483.
- Wang SM, Ho TS, Shen CFet al.Echovirus 18 meningitis in southern Taiwan. Pediatr Infect Dis J 2011;30: 259–260.
- Zhu Y, Zhou X, Liu Jet al.Molecular identification of human enteroviruses associated with aseptic meningitis in Yunnan province, Southwest China. Springerplus 2016;5: 1515.
- Oberste MS, Nix WA, Maher Ket al.Improved molecular identification of enteroviruses by RT-PCR and amplicon sequencing. J Clin Virol 2003;26: 375–377.
- Oberste MS, Maher K, Kilpatrick DRet al.Typing of human enteroviruses by partial sequencing of VP1. J Clin Microbiol 1999;37: 1288–1293.
- Tamura K, Peterson D, Peterson Net al.MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011;28: 2731–2739.
- Drummond AJ, Suchard MA, Xie Det al.Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 2012;29: 1969–1973.
- Lole KS, Bollinger RC, Paranjape RSet al.Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 1999;73: 152–160.
- Perera D, Yusof MA, Podin Yet al.Molecular phylogeny of modern coxsackievirus A16. Arch Virol 2007;152: 1201–1208.
- Reimann BY, Zell R, Kandolf R.Mapping of a neutralizing antigenic site of Coxsackievirus B4 by construction of an antigen chimera. J Virol 1991;65: 3475–3480.
- Chen XP, Sun SZ, Guo JYet al.Complete genome sequence analysis of echovirus 18 associated with aseptic meningitis in Hebei Province, China, in 2015. Genome Announc 2016;4: e01165–16.