
Abstract
Although several optimal mycobacterial interspersed repetitive units–variable number tandem repeat (MIRU–VNTR) loci have been suggested for genotyping homogenous Mycobacterium tuberculosis, including the Beijing genotype, a more efficient and convenient selection strategy for identifying optimal VNTR loci is needed. Here 281 M. tuberculosis isolates were analyzed. Beijing genotype and non-Beijing genotypes were identified, as well as Beijing sublineages, according to single nucleotide polymorphisms. A total of 22 MIRU–VNTR loci were used for genotyping. To efficiently select optimal MIRU–VNTR loci, we established accumulations of percentage differences (APDs) between the strains among the different genotypes. In addition, we constructed a minimum spanning tree for clustering analysis of the VNTR profiles. Our findings showed that eight MIRU–VNTR loci displayed disparities in h values of ≥0.2 between the Beijing genotype and non-Beijing genotype isolates. To efficiently discriminate Beijing and non-Beijing genotypes, an optimal VNTR set was established by adding loci with APDs ranging from 87.2% to 58.8%, resulting in the construction of a nine-locus set. We also found that QUB11a is a powerful locus for separating ST10s (including ST10, STF and STCH1) and ST22s (including ST22 and ST8) strains, whereas a combination of QUB11a, QUB4156, QUB18, Mtub21 and QUB26 could efficiently discriminate Beijing sublineages. Our findings suggested that two nine-locus sets were not only efficient for distinguishing the Beijing genotype from non-Beijing genotype strains, but were also suitable for sublineage genotyping with different discriminatory powers. These results indicate that APD represents a quantitative and efficient approach for selecting MIRU–VNTR loci to discriminate between divergent M. tuberculosis sublineages.
Emerging Microbes & Infections (2017) 6, e68; doi:10.1038/emi.2017.58; published online 26 July 2017
Introduction
The extensive spread of multidrug-resistant tuberculosis in Asia is predominantly driven by the rapid spread of the Mycobacterium tuberculosis Beijing genotype.Citation1, Citation2, Citation3, Citation4 Beijing genotype strains have been proposed to have an increased capacity to acquire drug resistance, have an increased transmission ability, exhibit hypervirulence and result in a more rapid progression to disease after infection.Citation5, Citation6, Citation7, Citation8, Citation9, Citation10 However, the association of infection with the Beijing strain with drug resistance and specific pathobiological or epidemiological characteristics has not been systematic.Citation11 Although this genotype is associated with drug resistance,Citation12, Citation13, Citation14, Citation15, Citation16 relatively weak associations have been reported in other geographic settings.Citation17, Citation18, Citation19 The heterogeneity suggests the existence of substantial intra-lineage biogeographical diversity affecting pathobiological properties.Citation20 The Beijing genotype includes several divergent sublineages,Citation21, Citation22, Citation23 the phenotypic differences among which have not been characterized. To date, it remains unclear whether the distribution of sublineages in different geographic areas is causally related to the variations in tuberculosis (TB) transmission dynamics and/or the prevalence of drug-resistant TB.
To explore the mechanisms associated with the spread of Mycobacterium spp. and to clarify the correlation between genotypes and phenotypes, extensive genotyping studies have been performed.Citation6, Citation20, Citation24, Citation25 In China, the prevalence of the Beijing genotype varies from 25% to 92% among geographic areas,Citation19 with the distribution of the Beijing sublineages also varying geographically.Citation20, Citation26, Citation27 This could lead to misunderstandings of the phenotypic features of M. tuberculosis strains and the Beijing genotype epidemic.
Over the previous decade, the molecular epidemiology of M. tuberculosis advanced markedly,Citation28, Citation29 mainly because of the development of variable number tandem repeat (VNTR) typing and its application to mycobacterial interspersed repetitive units (MIRUs).Citation30 Compared with IS6110-RFLP (insertion sequence 6110-restriction fragment length polymorphism) and spoligotyping, MIRU–VNTRs display a higher discriminatory power, especially in strains with low copy numbers of IS6110, and are suitable for long-term epidemiologic analyses because of their slightly slower evolution.Citation19, Citation28, Citation31, Citation32 VNTR genotyping is based on the discrimination power of a locus in a certain geographic region where a certain type of M. tuberculosis is dominant. However, there are geographic polymorphisms in certain VNTR loci, and even the standard 15- and 24-locus VNTR sets might not be optimal for discriminating M. tuberculosis isolates in geographic areas where the Beijing genotype strains are highly endemic.Citation33, Citation34, Citation35, Citation36 In addition, choosing a locus based only on its discriminatory power might result in genotyping errors for Beijing and non-Beijing family strains because of the different genetic features of the populations.
Single nucleotide polymorphisms (SNPs) have been used for genotyping sublineages of the Beijing family.Citation37, Citation38, Citation39, Citation40 An advantage of SNPs is that they allow the unambiguous assignment of a particular clone to a certain lineage; however, SNP typing is less informative in molecular epidemiology studies because of its low discriminatory power.Citation41
Luo et al.Citation42 conducted genotyping of Beijing strains by combining SNPs and VNTR, showing that SNPs in modern Beijing strains correctly classified clusters that were previously misclassified because of the limited discriminatory power and/or homoplasy of VNTR.Citation42 Therefore, the combinatorial use of VNTR and SNPs might allow discrimination of Beijing and non-Beijing strains, as well as sublineages of Beijing genotypes.
Table 1 Allelic diversity (h) of different MIRU–VNTR loci
Previously, we defined a 15-locus VNTR set to effectively distinguish M. tuberculosis isolates in Heilongjiang Province,Citation19 finding that Beijing family strains account for 90% of M. tuberculosis. In addition, there is a heavy burden of drug-resistant TB in Heilongjiang Province.Citation43 Accordingly, we focused on exploring genotype markers to distinguish Beijing and non-Beijing family isolates and developing appropriate genotyping approaches for areas with a high burden of Beijing genotype lineages. It is important to understand the possible mechanisms by which M. tuberculosis and drug-resistant TB spread; therefore, in this study, we established a new approach to evaluate the power of a VNTR locus and SNP genotyping for discriminating Beijing and non-Beijing family strains, as well as Beijing sublineages.
MATERIALS AND METHODS
Collection of M. tuberculosis clinical isolates and identification of the Beijing genotype and sublineages
This study included 281 M. tuberculosis samples isolated between May 2007 and October 2009 from 281 patients from various regions of Heilongjiang Province who had been diagnosed with pulmonary TB in the Harbin Chest Hospital.Citation27 Of these strains, 200 were described in our previous work,Citation19 with an additional 81 strains described here. The Beijing family strains were determined by RD105 deletion and the Rv0679c SNP.Citation27, Citation44 Non-Beijing family strains were identified by a long sequence polymorphism as described.Citation45 Sublineages of the Beijing family strains (sequence types (STs)) were previously determined by detection of nine SNPs that are only present in Beijing family strains.Citation27, Citation38 All primers used for polymerase chain reactions for determination of genotypes and sublineages are shown in Supplementary Table 1.
MIRU–VNTR typing
In this study, we used 22 MIRU–VNTR loci () for the genotyping of the 81 isolates. The 200 isolates previously genotyped by 15 VNTR loci were analyzed with seven new VNTR loci (VNTR3820, QUB3232, VNTR4120, QUB11a, QUB18, QUB4156 and Mtub24), resulting in the availability of 281 isolates with 22 VNTR loci for analysis.Citation19 MIRU–VNTR allelic diversity (h) at a given locus was calculated as follows: h=1−∑xi2 [n/(n−1)], where xi is the frequency of the ith allele at the locus, and n is the number of isolates.Citation46 Discrimination of the locus combination was calculated using the Hunter–Gaston discriminatory index (HGDI):Citation46
![]()
where N is the total number of isolates in the typing method, s is the number of distinct patterns discriminated by MIRU–VNTR and nj is the number of isolates belonging to the jth pattern.
Accumulation of the percentage differences (APDs) of the strains with the same repeat-number pattern at a certain VNTR locus between category A and category B were obtained as follows:
![]()
where s is the total number of distinct repeat patterns, Aj (Bj) is the percentage of A (B) category strains with the jth repeat pattern from all the strains of the A (B) category.
Minimum spanning tree (MST) construction and analysis of locus-set(s) discrimination
For classification of the strains of different genotypes based on a VNTR locus set, a minimum spanning tree (MST) was created using Bionumerics software version 6.6 (Applied Maths, St. Martens-Latem, Belgium) for the 281 M. tuberculosis strains based on VNTR genotyping and ST typing.Citation47 The rules for tree construction were the same as those previously reported.Citation47 An MST connects all the genetic profiles, such that the global genetic distance between all the branches is minimized.
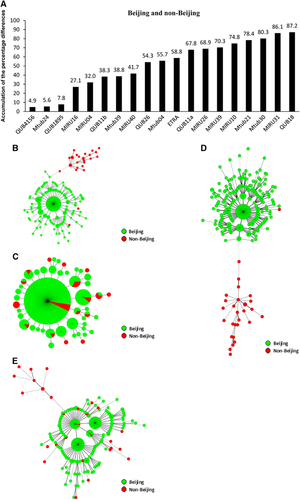
Figure 1 VNTR loci for discriminating the Beijing and non-Beijing family strains. The accumulations of the percentage differences of 19 VNTR loci between the Beijing and non-Beijing strains (A). MSTs of the 281 strains based on QUB18, MIRU31, Mtub30, MIRU10, MIRU39 and Mtub21 (B); QUB4156, Mtub24, QUB1895, MIRU16 and MIRU04 (C); QUB18, MIRU31, Mtub30, Mtub21, MIRU10, MIRU39, MIRU26, QUB11a and ETRA (D); and QUB4156, Mtub24, QUB1895, MIRU16, MIRU04, QUB11b, Mtub39 and MIRU40 (E). Beijing and non-Beijing strains are indicated in different colors.
RESULTS
Discriminatory power of the 22 VNTR loci for Beijing and non-Beijing isolates
A total of 281 M. tuberculosis isolates were analyzed, with 252 strains belonging to the Beijing genotype strains and 29 strains identified as the Euro-American genotype. We previously examined the discriminatory power of 15 VNTR loci and defined a combination of loci for genotyping M. tuberculosis isolates in Heilongjiang Province.Citation19 Here we examined additional VNTR loci to identify a more suitable set that exhibited better discriminatory power for genotyping local isolates. We first evaluated the discriminatory power of seven previously unused loci to investigate the local isolates, followed by re-evaluation of the 22 loci (15 previously evaluated and seven new) using a larger sample population (). Among the seven loci, three (VNTR3820, QUB3232 and VNTR4120) displayed an extremely high degree of discriminatory power for the Beijing and non-Beijing isolates in the study area. QUB11a and QUB18 showed moderate discrimination (0.3<h<0.6), whereas the other two loci, QUB4156 and Mtub24, showed low discrimination (h≤0.3).
We also found that some loci exhibited highly variable discrimination between Beijing and non-Beijing isolates. For example, eight loci displayed h-value disparities of ≥0.2 between the two isolate types. In addition, the loci QUB18, Mtub21 and MIRU40 displayed moderate or low discrimination values for Beijing isolates, but high discrimination values for non-Beijing isolates. Three VNTR loci (VNTR3820, QUB3232 and VNTR4120) were excluded from further analyses to avoid the influence of high allelic diversity on the genotyping results.
VNTR loci for discriminating Beijing and non-Beijing isolates
Because the VNTR loci exhibited different discriminatory powers between Beijing and non-Beijing isolates, we attempted to determine the optimal VNTR loci for this process. Given that there are differences in allelic diversity (h) and repeat number at some VNTR loci between Beijing and non-Beijing isolates (Supplementary Figure S1), we hypothesized that the difference, specifically the accumulative difference in allelic diversity, represents the discriminative capacity of the locus for various isolate types. Therefore, we calculated the APD for each locus as described in the ‘Materials and Methods’ section.
To confirm our hypothesis, we constructed an MST using all M. tuberculosis isolates. We found that the Beijing and non-Beijing isolates were separated into two main populations when we used QUB18, MIRU31, Mtub30, MIRU10, MIRU39 and Mtub21, with APD values >70% (). MST analysis indicated that 93.1% (27/29) of non-Beijing isolates belonged to one branch, whereas 100% (252/252) of Beijing isolates belonged to another branch (). The APDs of the locus set, including the other five loci (QUB4156, Mtub24, QUB1895, MIRU16 and MIRU04), were low (4.9%–32.0%). These results indicated that the set exhibited a far worse capacity for discrimination relative to that of the former set ().
Similar results were obtained using APD values for seven-, eight-, and nine-locus sets. For example, we chose nine loci based on the descending order of the APD values. The APDs, arranged from high to low for the addition of each locus (that is, QUB18, MIRU31, Mtub30, Mtub21, MIRU10, MIRU39, MIRU26, QUB11a and ETRA), ranged from 87.2% to 58.8%. In addition, MST analysis indicated the same two branches, with 96.6% of the non-Beijing isolates found on one branch (). However, when we chose loci based on the APD values in ascending order, the MST for the loci (QUB4156, Mtbu24, QUB1895, MIRU16, MIRU04, QUB11b, Mtub39 and MIRU40) with APDs ranging from 4.9% to 41.7% did not indicate clusters of Beijing or non-Beijing isolates and failed to classify isolates of each genotype to specific MST branches (). We ultimately determined that the VNTR loci with APDs >56% could efficiently discriminate the Beijing and non-Beijing isolates.
VNTR loci for deep discrimination of Beijing sublineages
We previously found that Beijing genotype isolates account for ~90% of all M. tuberculosis clinical isolates and that ST10 was dominant (63.2%) among the 10 STs found in the study area.Citation27 Here we combined ST and VNTR genotyping to precisely classify clinical isolates and identify appropriate VNTR loci for areas where Beijing genotype isolates are dominant.
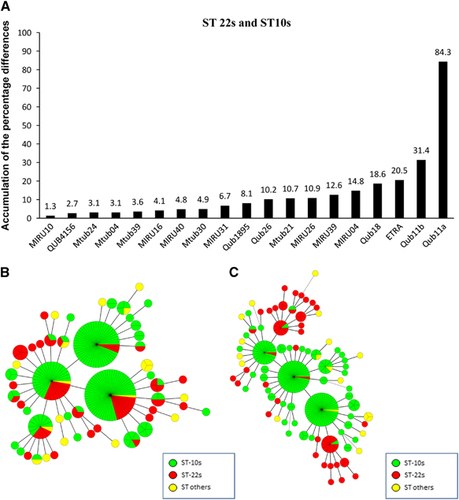
We first calculated the frequencies of strains with a certain repeat number at a particular VNTR locus to classify the ST strains (Supplementary Figure S2). All the Beijing isolates were grouped into three ST categories: ST10s (173 isolates, including ST10, STF and STCH1), ST22s (61 isolates, including ST22 and ST8) and ST others (18 isolates, including ST25, ST26, ST11, ST19 and ST3) (Supplementary Table S2). We then compared the pairwise difference in APDs of the VNTR loci between ST categories (ST10s vs ST22s, ST22s vs ST others and ST10s vs ST others) (,). We found that QUB11a was the most powerful locus, with an APD of 84.3% and high level of discrimination between ST10s and ST22s strains (). The APDs for the other VNTR loci were from 1.3% to 31.4% and exhibited lower levels of discrimination for different ST categories.
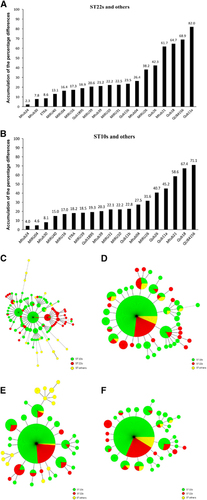
To confirm the power of QUB11a and the methodology for locus selection, we chose additional loci based on APDs in descending order. For example, QUB11b, ETRA and QUB18 showed APDs in the range of 31.4% to 18.6%. The MST did not indicate clustering of the strains from the same sublineages and failed to assign strains of each sublineage to a specific MST branch (). Using QUB11a, we observed that ST10s and ST22s strains formed distinct groups, with ST10s strains divided into three clusters and ST22s strains belonging to two separate clusters (). Using the same strategy, we found five loci capable of discriminating ST22s and ST others within the study area (that is, QUB11a, QUB4156, QUB18, Mtub21 and QUB26; APDs: 82.0%–42.3%) (). By contrast, Mtub24, ETRA, Mtub30, MIRU40 and MIRU04 were unable to discriminate these two groups (). These findings indicated that loci with APDs >40% efficiently discriminated ST22s strains from ST others strains.
The APD order for loci capable of discriminating ST10s strains from ST others strains was similar with that of ST22s strains, except for QUB11a (). We found three loci capable of discriminating ST10s from ST others (that is, QUB4156, QUB18 and Mtub21; APDs: 71.1%–58.6%) (). As expected, the four-locus set (Mtub24, MIRU04, Mtub30 and MIRU40) exhibiting low APDs did not distinguish ST10s strains from ST others strains ().
We concluded that VNTR loci used to discriminate dominant Beijing genotype sublineages in the study area should have APDs >40%. QUB11a could discriminate ST22s strains from ST10s strains, whereas three loci (QUB4156, QUB18 and Mtub21) were able to discriminate ST others strains from ST10s or ST22s strains. Furthermore, addition of QUB26 did not influence the discrimination of Beijing genotype sublineages (data not shown) but did increase the discriminatory power. Therefore, a combination of QUB11a, QUB4156, QUB18, Mtub21 and QUB26 was used as the first-line set for genotyping the Beijing sublineages.
Figure 2 VNTR loci for discriminating between the ST10s and ST22s strains of the Beijing family. The accumulation of the percentage differences of the 19 VNTR loci between the ST10s and ST22s strains (A). MSTs of the 252 Beijing strains based on QUB11b, ETRA and QUB18 (B); QUB11a, QUB11b, ETRA and QUB18 (C). ST22s, ST10s and ST others strains are indicated in different colors.
Genotyping loci for discriminating locally endemic sublineages
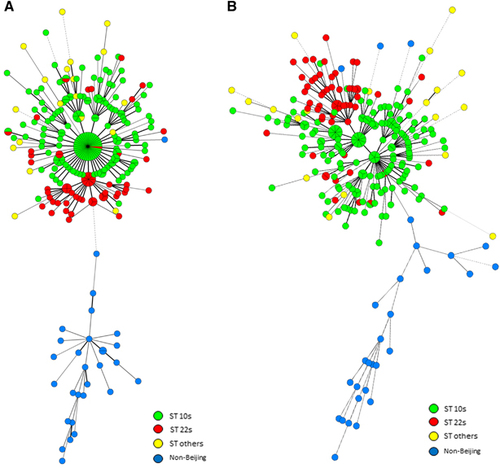
According to the locus selection strategy described here and the loci exhibiting discriminatory power for local sublineages, we found that the nine-locus combination with the top APDs between Beijing and non-Beijing genotype strains classified all the strains into two clusters, with 100% of the Beijing strains belonging to one branch and 96.6% of the non-Beijing strains belonging to another branch (HGDI: 0.979; cluster rate: 30%) (), and that the ST22s and ST10s strains were effectively divided. Another nine-locus combination with the highest APDs among the sublineages of Beijing genotype strains also discriminated Beijing from non-Beijing strains (). In this analysis, 89.7% of the non-Beijing strains were found on one branch and the ST22s strains accounted for 88% of the ST22s branch (HGDI and cluster rate: 0.997 and 14.5%, respectively).
DISCUSSION
MIRU–VNTR genotyping using the standardized 24-locus set has become an international standard and is currently applied for M. tuberculosis genotyping in China and worldwide.Citation48 Both 12-locus and 15-locus sets have been proposed as alternatives for rapid and high-throughput genotyping,Citation49 with a recent study indicating that a 15-locus VNTR set could be very useful for the genotyping of M. tuberculosis complex strains in China and that a new 19-locus set was informative and could be useful for epidemiological studies of Beijing genotype strains.Citation50
However, accounting for strain polymorphism, geographical differences in isolate distribution and variations in the prevalence of Beijing strains in China, the allelic diversity (h) of a certain locus varies, with these loci potentially exhibiting different discriminatory powers.Citation19 Of the standard 24-locus set, MIRU20, Mtub34, MIRU02 and MIRU24 were excluded from the present study because of their low rates of polymorphism and h values <0.1 in Asia.Citation51 As reported previously, QUB11a and QUB18, which are also not included in the 24-standard loci, display moderate discriminatory power in Heilongjiang Province and are informative and optimal for M. tuberculosis genotyping in this area.Citation19 Because it is accepted that loci with either extremely high (h>0.8) or low (h<0.1) allelic diversity are unsuitable for genotyping because of their variation or low discriminatory power, the three loci exhibiting extremely high allelic diversity (VNTR3820, QUB3232 and VNTR4120) in M. tuberculosis isolates from the region should also be omitted. In this study, even within a single region (that is, Heilongjiang Province), we found that the allelic diversity at a particular locus differed between Beijing and non-Beijing strains. Therefore, we analyzed the remaining loci to identify the VNTR locus or combination of loci that was capable of discriminating Beijing- from non-Beijing genotype strains.
Figure 3 VNTR loci for discriminating ST22s or ST10s strains from ST others strains of the Beijing family. The accumulation of the percentage differences of 19 VNTR loci between ST10s and ST others strains (A) and between ST22s and ST others strains (B). MSTs of the 252 Beijing strains based on QUB11a, QUB4156, QUB18, Mtub21 and QUB26 (C); Mtub24, ETRA, Mtub30, MIRU40 and MIRU04 (D); QUB4156, QUB18 and Mtub21 (E); and Mtub24, MIRU04, Mtub30 and MIRU40 (F). ST22s, ST10s, and ST others strains are indicated in different colors.
The combination of MIRU26, MIRU31 and ETRA was established for the differentiation of Beijing-lineage isolates, with a sensitivity of 94.7% and a specificity of 98.5%.Citation52 A previous study showed that Mtub30 and Mtub02 can serve as alternatives for identification of Beijing genotype stains in resource-limited regions.Citation53 Compared with genetically divergent non-W-Beijing strains, Beijing genotype strains contained a significantly higher number of tandem repeats in the loci (ETRA, ETR-E, QUB26, QUB18, QUB11B and Mtub21). Furthermore, W-Beijing genomes frequently possess seven, eight or nine tandem repeats at loci QUB26 and QUB18, indicating a pattern that is different from other genotypes.Citation54
The above-mentioned studies focused on difference in the number of repeats among all strains at a given locus and their contribution to the differentiation of the Beijing strains. The proportions of each Beijing sublineage categorized according to the repeat number at a particular VNTR locus were applied for the selection of phylogenetically informative VNTR loci based on the dominant percentages of genotypes that exhibited different repeat-number patterns.Citation40, Citation55, Citation56 This method is based on the repeat-number differences and is less quantitative, whereas another method for choosing a locus combination is based on the highest cumulative HGDI accompanied by successive addition of each MIRU–VNTR locus according to the allele diversity.Citation57 As first-line VNTR locus sets for genotyping M. tuberculosis, loci with extremely high and low allele diversity should be excluded;Citation36 however, some alternative loci might also exhibit the same allele diversity, resulting in difficulties in choosing suitable loci. Furthermore, considering the discriminatory redundancy between loci, we need to try many combinations to obtain the optimal set and the discriminatory power for genotyping M. tuberculosis genotypes may be neglected.Citation36 In this study, we adopted a new method that considers the APD of a VNTR locus to distinguish the Beijing from non-Beijing strains. This approach is based on the proportional difference between strains having the same repeat number at a certain VNTR locus between Beijing and non-Beijing strains and the accumulative difference of all the repeat patterns. In this method, a locus set is selected according to the APD values in descending order, facilitating the selection of better locus sets for discriminating Beijing and non-Beijing strains.
To classify Beijing sublineages based on the VNTR loci, the proportions of each Beijing sublineage defined by SNPs were analyzed based on the number of repeats at a certain VNTR locus. Ancient and modern Beijing sublineages can be classified by different VNTR loci from different regions (for example, MIRU31 and Mtub04 in Taiwan; Mtub21 and QUB4156 in Japan, Thailand and Peru).Citation40, Citation55, Citation56, Citation58 Furthermore, STs of Beijing sublineages defined by SNPs can be distinguished based on the repeat number at a VNTR locus. For example, MIRU10, QUB11b and VNTR3155 display unique repeat numbers in ST3, STK and ST26 strains, respectively.Citation58
In our previous study, we found that locally prevalent M. tuberculosis strains were modern STs, including ST22 and ST10,Citation27 and that some sublineages are genetically close to either ST22 or ST10, whereas ancient sublineages are less dominant. Therefore, in this study, we divided all sublineages into three groups: ST22s, ST10s and ST others (ancient sublineages). Application of APDs to select VNTR loci for the differentiation of Beijing sublineages revealed that this method was efficient for VNTR locus selection. We found that only one locus (QUB11a) was sufficient for differentiating ST10s from ST22s strains, with an APD of 84.3%, whereas the other loci showed lower discriminatory power, with APDs <32%. Five loci (QUB11a, QUB4156, QUB18, Mtub21 and QUB26) were more effective at discriminating ST22s from ST others strains, with APDs ranging from 82.0% to 42.3%. In addition, the five-locus combination showed stronger discriminatory power relative to that observed from using individual loci, and three loci (QUB4156, QUB18 and Mtub21), with APDs ranging from 71.1% to 58.6%, effectively classified strains as ST10s or ST others. Our results suggested that the APD method described here will help to more efficiently select (sub)lineage-discriminating VNTR loci.
Figure 4 MSTs of the 281 strains based on QUB11a, QUB18, Mtub21, MIRU26, MIRU31, MIRU39, ETRA, MIRU10 and Mtub30 (A); and QUB11a, QUB18, QUB4156, Mtub21, QUB26, MIRU26, QUB11b, MIRU31 and Mtub04 (B). Non-Beijing family and sublineages of the Beijing family strains are indicated in different colors.
In summary, we found that the APD allowed effective determination of the VNTR loci for typing M. tuberculosis into different genotypes or sublineages. Possible optimal VNTR locus sets should include VNTR loci with high APD values between the two targeted groups because of the ability of the APD to increase the discriminatory power of the VNTR locus set. The limitation of the present work is that our VNTR loci were selected according to known and previously identified loci. In future work, we will examine additional loci that might exhibit higher discriminatory power and that could be suitable for discriminating M. tuberculosis isolates from various areas of the world.
Supplementary Table S2
Download MS Word (18 KB)Supplementary Table S1
Download PDF (20.3 KB)Supplementary Figure S1
Download PDF (121.1 KB)Supplementary Figure S2
Download PDF (69 KB)Acknowledgments
This work was supported by the Grant for Innovation Team Program of Heilongjiang Province to HL and DL. This work was also supported by research funding for the Key Laboratory of Heilongjiang Province College Medical Etiology (BSL3). This work was partially supported by the Research Program on Emerging and Re-emerging Infectious Diseases from the Japan Agency for Medical Research and development, AMED.
Supplementary Information for this article can be found on the Emerging Microbes & Infections website (http://www.nature.com/emi)
Related Research Data
References
- Niemann S, Diel R, Khechinashvili Get al.Mycobacterium tuberculosis Beijing lineage favors the spread of multidrug-resistant tuberculosis in the Republic of Georgia. J Clin Microbiol 2010;48: 3544–3550.
- Stoffels K, Allix-Beguec C, Groenen Get al.From multidrug- to extensively drug-resistant tuberculosis: upward trends as seen from a 15-year nationwide study. PLoS ONE 2013;8: e63128.
- Casali N, Nikolayevskyy V, Balabanova Yet al.Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet 2014;46: 279–286.
- Munsiff SS, Nivin B, Sacajiu Get al.Persistence of a highly resistant strain of tuberculosis in New York City during 1990-1999. J Infect Dis 2003;188: 356–363.
- Glynn JR, Whiteley J, Bifani PJet al.Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg Infect Dis 2002;8: 843–849.
- Merker M, Blin C, Mona Set al.Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet 2015;47: 242–249.
- Bifani PJ, Mathema B, Kurepina NEet al.Global dissemination of the Mycobacterium tuberculosis W-Beijing family strains. Trends Microbiol 2002;10: 45–52.
- Parwati I, van Crevel R, van Soolingen D.Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. Lancet Infect Dis 2010;10: 103–111.
- Hanekom M, Gey van Pittius NC, McEvoy Cet al.Mycobacterium tuberculosis Beijing genotype: a template for success. Tuberculosis 2011;91: 510–523.
- de Jong BC, Hill PC, Aiken Aet al.Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis lineage in the Gambia. J Infect Dis 2008;198: 1037–1043.
- Kato-Maeda M, Shanley CA, Ackart Det al.Beijing sublineages of Mycobacterium tuberculosis differ in pathogenicity in the guinea pig. Clin Vaccine Immunol 2012;19: 1227–1237.
- Zhang Z, Lu J, Liu Met al.Genotyping and molecular characteristics of multidrug-resistant Mycobacterium tuberculosis isolates from China. J Infect 2015;70: 335–345.
- Kozinska M, Augustynowicz-Kopec E.Drug resistance and population structure of Mycobacterium tuberculosis Beijing strains isolated in Poland. Pol J Microbiol 2015;64: 399–401.
- Pang Y, Zhou Y, Zhao Bet al.Spoligotyping and drug resistance analysis of Mycobacterium tuberculosis strains from national survey in China. PLoS One 2012;7: e32976.
- Zhang Z, Pang Y, Wang Yet al.Beijing genotype of Mycobacterium tuberculosis is significantly associated with linezolid resistance in multidrug-resistant and extensively drug-resistant tuberculosis in China. Int J Antimicrob Agents 2014;43: 231–235.
- Liu BB, Lu LP, Lu Bet al.Meta analysis on the correlation between Mycobacterium tuberculosis Beijing family strains and drug resistance. Zhonghua Yu Fang Yi Xue Za Zhi 2012;46: 158–164.
- Jou R, Chiang CY, Huang WL.Distribution of the Beijing family genotypes of Mycobacterium tuberculosis in Taiwan. J Clin Microbiol 2005;43: 95–100.
- Mathuria JP, Srivastava GN, Sharma Pet al.Prevalence of Mycobacterium tuberculosis Beijing genotype and its association with drug resistance in North India. J Infect Public Health 2017;10: 409–414.
- Wang J, Liu Y, Zhang CLet al.Genotypes and characteristics of clustering and drug susceptibility of Mycobacterium tuberculosis isolates collected in Heilongjiang Province, China. J Clin Microbiol 2011;49: 1354–1362.
- Mokrousov I.Mycobacterium tuberculosis phylogeography in the context of human migration and pathogen's pathobiology: insights from Beijing and Ural families. Tuberculosis 2015;95 (Suppl 1): S167–S176.
- Garzelli C, Lari N, Rindi L.Genomic diversity of Mycobacterium tuberculosis Beijing strains isolated in Tuscany, Italy, based on large sequence deletions, SNPs in putative DNA repair genes and MIRU-VNTR polymorphisms. Tuberculosis 2016;97: 147–153.
- Liu Q, Luo T, Dong Xet al.Genetic features of Mycobacterium tuberculosis modern Beijing sublineage. Emerg Microbes Infect 2016;5: e14.
- Rindi L, Lari N, Cuccu Bet al.Evolutionary pathway of the Beijing lineage of Mycobacterium tuberculosis based on genomic deletions and mutT genes polymorphisms. Infect Genet Evol 2009;9: 48–53.
- Coscolla M, Gagneux S.Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol 2014;26: 431–444.
- Warner DF, Koch A, Mizrahi V.Diversity and disease pathogenesis in Mycobacterium tuberculosis. Trends Microbiol 2015;23: 14–21.
- Chaidir L, Sengstake S, de Beer Jet al.Predominance of modern Mycobacterium tuberculosis strains and active transmission of Beijing sublineage in Jayapura, Indonesia Papua. Infect Genet Evol 2016;39: 187–193.
- Li D, Dong CB, Cui JYet al.Dominant modern sublineages and a new modern sublineage of Mycobacterium tuberculosis Beijing family clinical isolates in Heilongjiang Province, China. Infect Genet Evol 2014;27: 294–299.
- Ei PW, Aung WW, Lee JSet al.Molecular strain typing of Mycobacterium tuberculosis: a review of frequently used methods. J Korean Med Sci 2016;31: 1673–1683.
- Witney AA, Cosgrove CA, Arnold Aet al.Clinical use of whole genome sequencing for Mycobacterium tuberculosis. BMC Med 2016;14: 46.
- Kanduma E, McHugh TD, Gillespie SH.Molecular methods for Mycobacterium tuberculosis strain typing: a users guide. J Appl Microbiol 2003;94: 781–791.
- Hachisu Y, Hashimoto R, Kishida Ket al.Analytical procedure of variable number of tandem repeats (VNTR) analysis and effective use of analysis results for tuberculosis control. Rinsho Byori 2013;61: 1123–1135.
- Mears J, Abubakar I, Cohen Tet al.Effect of study design and setting on tuberculosis clustering estimates using mycobacterial interspersed repetitive units-variable number tandem repeats (MIRU-VNTR): a systematic review. BMJ Open 2015;5: e005636.
- Alonso-Rodriguez N, Martinez-Lirola M, Sanchez MLet al.Prospective universal application of mycobacterial interspersed repetitive-unit-variable-number tandem-repeat genotyping to characterize Mycobacterium tuberculosis isolates for fast identification of clustered and orphan cases. J Clin Microbiol 2009;47: 2026–2032.
- de Beer JL, Kremer K, Kodmon Cet al.First worldwide proficiency study on variable-number tandem-repeat typing of Mycobacterium tuberculosis complex strains. J Clin Microbiol 2012;50: 662–669.
- Streit E, Baboolal S, Akpaka PEet al.Finer characterization of Mycobacterium tuberculosis using spoligotyping and 15-loci MIRU-VNTRs reveals phylogeographical specificities of isolates circulating in Guyana and Suriname. Infect Genet Evol 2015;30: 114–119.
- Luo T, Yang C, Pang Yet al.Development of a hierarchical variable-number tandem repeat typing scheme for Mycobacterium tuberculosis in China. PLoS One 2014;9: e89726.
- Srilohasin P, Chaiprasert A, Tokunaga Ket al.Genetic diversity and dynamic distribution of Mycobacterium tuberculosis isolates causing pulmonary and extrapulmonary tuberculosis in Thailand. J Clin Microbiol 2014;52: 4267–4274.
- Filliol I, Motiwala AS, Cavatore Met al.Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol 2006;188: 759–772.
- Liu Y, Wang S, Lu Het al.Genetic diversity of the Mycobacterium tuberculosis Beijing family based on multiple genotyping profiles. Epidemiol Infect 2016;144: 1728–1735.
- Chen YY, Chang JR, Huang WFet al.Genetic diversity of the Mycobacterium tuberculosis Beijing family based on SNP and VNTR typing profiles in Asian countries. PLoS One 2012;7: e39792.
- Cardoso Oelemann M, Gomes HM, Willery Eet al.The forest behind the tree: phylogenetic exploration of a dominant Mycobacterium tuberculosis strain lineage from a high tuberculosis burden country. PLoS ONE 2011;6: e18256.
- Luo T, Yang C, Gagneux Set al.Combination of single nucleotide polymorphism and variable-number tandem repeats for genotyping a homogenous population of Mycobacterium tuberculosis Beijing strains in China. J Clin Microbiol 2012;50: 633–639.
- Li D, Wang JL, Ji BYet al.Persistently high prevalence of primary resistance and multidrug resistance of tuberculosis in Heilongjiang Province, China. BMC Infect Dis 2016;16: 516.
- Nakajima C, Tamaru A, Rahim Zet al.Simple multiplex PCR assay for identification of Beijing family Mycobacterium tuberculosis isolates with a lineage-specific mutation in Rv0679c. J Clin Microbiol 2013;51: 2025–2032.
- Gagneux S, Small PM.Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis 2007;7: 328–337.
- Selander RK, Caugant DA, Ochman Het al.Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl Environ Microbiol 1986;51: 873–884.
- Wada T, Iwamoto T, Maeda S.Genetic diversity of the Mycobacterium tuberculosis Beijing family in East Asia revealed through refined population structure analysis. FEMS Microbiol Lett 2009;291: 35–43.
- Supply P, Allix C, Lesjean Set al.Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol 2006;44: 4498–4510.
- Mazars E, Lesjean S, Banuls ALet al.High-resolution minisatellite-based typing as a portable approach to global analysis of Mycobacterium tuberculosis molecular epidemiology. Proc Natl Acad Sci USA 2001;98: 1901–1906.
- Jiang Y, Liu HC, Zheng Het al.19-VNTR loci used in genotyping Chinese clinical Mycobacterium tuberculosis complex strains and in association with spoligotyping. J Basic Microbiol 2013;53: 562–580.
- Mokrousov I, Narvskaya O, Vyazovaya Aet al.Mycobacterium tuberculosis Beijing genotype in Russia: in search of informative variable-number tandem-repeat loci. J Clin Microbiol 2008;46: 3576–3584.
- Chin PJ, Chiu CC, Jou R.Identification of Beijing lineage Mycobacterium tuberculosis with combined mycobacterial interspersed repetitive unit loci 26, 31, and ETR-A. J Clin Microbiol 2007;45: 1022–1023.
- Liu R, Xing L, Peng Zet al.Usefulness of mycobacterial interspersed repetitive-unit locus PCR amplification in rapid diagnosis of Beijing lineage strain infection among pediatric tuberculosis patients. J Clin Microbiol 2011;49: 712–714.
- Surikova OV, Voitech DS, Kuzmicheva Get al.Efficient differentiation of Mycobacterium tuberculosis strains of the W-Beijing family from Russia using highly polymorphic VNTR loci. Eur J Epidemiol 2005;20: 963–974.
- Faksri K, Drobniewski F, Nikolayevskyy Vet al.Genetic diversity of the Mycobacterium tuberculosis Beijing family based on IS6110, SNP, LSP and VNTR profiles from Thailand. Infect Genet Evol 2011;11: 1142–1149.
- Iwamoto T, Grandjean L, Arikawa Ket al.Genetic diversity and transmission characteristics of Beijing family strains of Mycobacterium tuberculosis in Peru. PLoS ONE 2012;7: e49651.
- Kam KM, Yip CW, Tse LWet al.Optimization of variable number tandem repeat typing set for differentiating Mycobacterium tuberculosis strains in the Beijing family. FEMS Microbiol Lett 2006;256: 258–265.
- Wada T, Iwamoto T.Allelic diversity of variable number of tandem repeats provides phylogenetic clues regarding the Mycobacterium tuberculosis Beijing family. Infect Genet Evol 2009;9: 921–926.