
Abstract
Human adenovirus type 14 (HAdV-B14p) was originally identified as an acute respiratory disease (ARD) pathogen in The Netherlands in 1955. For approximately fifty years, few sporadic infections were observed. In 2005, HAdV-B14p1, a genomic variant, re-emerged and was associated with several large ARD outbreaks across the U.S. and, subsequently, in Canada, the U.K., Ireland, and China. This strain was associated with an unusually higher fatality rate than previously reported for both this prototype and other HAdV types in general. In China, HAdV-B14 was first observed in 2010, when two unrelated HAdV-B14-associated ARD cases were reported in Southern China (GZ01) and Northern China (BJ430), followed by three subsequent outbreaks. While comparative genomic analysis, including indel analysis, shows that the three China isolates, with whole genome data available, are similar to the de Wit prototype, all are divergent from the U.S. strain (303600; 2007). Although the genomes of strains GZ01 and BJ430 are nearly identical, as per their genome type characterization and percent identities, they are subtly divergent in their genome mutation patterns. These genomes indicate possibly two lineages of HAdV-B14 and independent introductions into China from abroad, or subsequent divergence from one; CHN2012 likely represents a separate sub-lineage. Observations of these simultaneously reported emergent strains in China add to the understanding of the circulation, epidemiology, and evolution of these HAdV pathogens, as well as provide a foundation for developing effective vaccines and public health strategies, including nationwide surveillance in anticipation of larger outbreaks with potentially higher fatality rates associated with HAdV-B14p1.
Emerging Microbes & Infections (2017) 6, e92; doi:10.1038/emi.2017.78; published online 1 November 2017
Introduction
Adenoviruses are human pathogens, causing infections and diseases of the respiratory, ocular, gastrointestinal, genitourinary, and metabolic systems, including being linked to obesity.Citation1 Interestingly, human adenovirus (HAdV) isolates with highly similar genomes are associated with sporadic and limited infections as well as with larger, wide-spread outbreaks, particularly in environments with close-quartered and/or vulnerable populations.Citation2, Citation3, Citation4, Citation5, Citation6, Citation7, Citation8, Citation9, Citation10, Citation11, Citation12, Citation13, Citation14 In terms of public health, although HAdV infections may be highly contagious, they are generally associated with high morbidity rates rather than high mortality rates;Citation13, Citation15, Citation16 therefore, HAdVs do not command the attention shown to more deadly viral pathogens such as HIV, Ebola virus, and SARS, and MERS coronaviruses. However, in children and immunocompromised individuals, particularly organ transplant recipients, HAdV infections may lead to significant rates of mortality.Citation13, Citation17, Citation18, Citation19
To date, applying high-resolution genomic and bioinformatic approaches has yielded in-depth and clearer views of the natural variation of HAdVs.Citation9, Citation12, Citation14, Citation20, Citation21, Citation22, Citation23, Citation24, Citation25, Citation26, Citation27, Citation28, Citation29, Citation30, Citation31, Citation32, Citation33, Citation34, Citation35, Citation36, Citation37 In particular, the molecular evolution and mechanisms of the genesis of emergent HAdV pathogens are being revealed; for example, recombination appears to be a major driving forceCitation14, Citation20, Citation21, Citation22, Citation23, Citation25, Citation26, Citation29, Citation38, Citation39 and zoonosis has been revealed as the origin for at least one important HAdV respiratory pathogen.Citation36, Citation37, Citation40 Computational analysis of the genomes from these emergent HAdV pathogens has led to laboratory-based experiments to explore consequences of specific mutations and their roles in pathogenic properties;Citation22, Citation27, Citation29, Citation41 for example, examining the nonsynonymous mutation in the fiber gene that potentially affects host receptor binding, which may contribute to the robustness and altered pathogenic properties of the re-emergent HAdV-14p1.Citation41
More than 84 genotypes, including all previously characterized serotypes, have been sequenced, characterized, and classified within seven species A–G (http://hadvwg.gmu.edu/).Citation21, Citation24, Citation28, Citation29, Citation30, Citation31, Citation42 Of these, species B, C, and E viruses are associated with respiratory diseases.Citation17, Citation43 Furthermore, species B HAdVs have been divided into subspecies B1 and B2, based on their genome similarities and restriction enzyme digestion patterns,Citation44 as well as reports of viruses of these two subspecies showing different tissue tropism.Citation45 Members of subspecies B1, i.e., HAdV-B3, -B7, -B16, and -B21, are respiratory pathogens, presumably due to their ability to infect cells of the respiratory tract; HAdV-B50 is also a subspecies B1 virus but has not been associated with a specific, if any, disease.Citation46 Subspecies B2 comprises HAdV-B11, -B14, -B34, -B35, and -B55; of these, HAdV-B11, -B34 and -B35 are recognized as renal or urinary tract pathogens.Citation17 The remaining two, HAdV-B14 and -B55, are respiratory tract pathogens.Citation2, Citation14 HAdV-B55 is a ‘Trojan Horse’ recombinant with a genome that is essentially that of a respiratory pathogen, HAdV-B14, but with the serologic appearance of a renal pathogen.Citation6, Citation7, Citation8, Citation9, Citation10, Citation14 HAdV-B14 was originally identified in a military trainee population as an acute respiratory disease (ARD) pathogen in The Netherlands in 1955,Citation2 with a second occurrence reported in a civilian setting in England in the same year (1955).Citation3 Since then and over the course of approximately fifty years, aside from a few sporadic and limited infections, HAdV-B14 has rarely been reported.
Beginning in 2005, HAdV-B14 re-emerged in the U.S., with multiple outbreaks in both civilian and military settings, as a highly contagious respiratory pathogen that was associated with a 76% hospitalization rate and an 18% fatality rate.Citation47, Citation48, Citation49 This fatality rate was alarming and unusual for a HAdV epidemic, particularly in presumably otherwise healthy adults. The same genome type, HAdV-B14p1, appeared in Europe with two ARD outbreaks in Ireland (2009) and the U.K. (2011).Citation5, Citation11, Citation50 These resulted in the highest fatality rates reported for HAdV-B14 outbreaks, at 33% and 23%, respectively.Citation5, Citation11, Citation50 In 2011, this pathogen re-emerged in Canada and caused one death in the three patients hospitalized with ARD.Citation49 In China, there were no HAdV-B14 cases reported until 2010, when HAdV-B14 emerged nearly simultaneously in two geographically distinct locations (Guangzhou and Beijing).Citation12, Citation32 Following these two reports, at least three additional HAdV-B14-related ARD outbreaks in China were recorded: One occurred in the Gansu Province (2011) in which 43 students in an elementary school setting presented with febrile respiratory illnesses,Citation51 one occurred in Beijing (2012) in which 30 adults presented with severe symptoms that required hospitalization,Citation33 and the third occurred in Liaoning Province (2012) in which 24 students in a middle school presented with febrile respiratory illnesses.Citation52 Unlike the U.S. 303600 strain, these five China strains did not have fatalities associated with them.
Given the severity of the symptoms and the numbers of afflicted individuals in China, the earlier outbreaks, with associated higher morbidity and fatality rates in the U.S., Canada, and Europe, and the putative transmission of HAdV-B14 from U.S. to Asia (South Korea) by military trainees (2007),Citation53 it is critical to determine the relationships between these China isolates and their global counterparts in order using high resolution genome analysis to understand the epidemiology of this re-emergent pathogen. These data will provide a basis for public health preventive measures, including diagnosis, surveillance, and appropriate protocols for limiting outbreaks and prevention. Specific information of the mutation rates, mutation hotspots, and new variations, particularly at their serologic recognition sites, will play a key role in the development of vaccines against these pathogens. This report presents the bioinformatics analysis of the HAdV-B14p1 Guangzhou strain (GZ01; October 2010), which was the first HAdV-B14 strain isolated in China,Citation12 and compares it with the contemporaneous Beijing strain (BJ430; December 2010) as well as a subsequent strain isolated in 2012 (CHN2012; February 2012). Provided is a high-resolution view of highly similar yet intriguingly divergent genomes, linking one of these strains with the globally re-emergent strain isolated in the U.S. in 2005, and suggesting at least two co-circulating lineages of type 14.
Materials and methods
Cells, virus stock, and DNA preparation
HAdV-B14p1 GZ01was isolated from a throat swab of a 17-month-old child hospitalized with acute suppurative tonsillitis in Guangzhou, China (October 2010).Citation12 Viral DNA was extracted using a Viral DNA Extraction Kit (Omega Bio-Tek Inc Corp; Norcross, GA). The protocol for PCR amplification is described earlier.Citation54 All the experimental protocols in this study were approved by the institutional ethics committee of Southern Medical University and were carried out in accordance with the approved guidelines. The informed consent for participation in this study was obtained from the guardian of the under-aged participant. Data records of the sample and sample collection are de-identified and completely anonymous.
Genome sequencing and annotation
The genome sequence of HAdV-B14p1 strain GZ01 was obtained using the Sanger sequencing method following PCR amplification of targeted overlapping 1–2 kb regions that covered the entire genome, as described earlier.Citation12 The sequence data, collected with an ABI 3730 Genetic Analyzer, provided an average genome coverage of 3 to 5 folds, with both strands represented. Gaps and ambiguous sequences were PCR-amplified using different primers and resequenced. DNA sequence fragments were assembled using the SEQMAN software from the Lasergene package (DNAStar; Madison, WI) into a single contig. The genome was annotated with an annotation protocol used for the HAdV-C1 genome analysis,Citation35 by first dividing the sequence into contiguous 1 kb non-overlapping segments.
Sequence analysis
Comparison of sequence differences between and spanning the genomes was performed with Genome Mutation Mapper (GMM; unpublished) software developed by the authors. GMM compares two or more genomes for nucleotide differences, noting SNPs and indels of query genomes relative to a reference; the data were confirmed with DNA Sequencher v5.1 (GeneCodes, Inc.; Ann Arbor, MI). Additionally, pairs of genome sequences were aligned using EMBOSS (http://www.ebi.ac.uk/Tools/emboss/) to show sequence identity, which provided a visualization of the alignment. Nucleic acid and amino acid sequence percent identities were calculated using software which was part of the EMBOSS package. Pair-wise comparisons of genomes were performed with the LAGAN (Limited Area Global Alignment of Nucleotides) program of mVISTA (http://genome.lbl.gov/vista/lagan/submit.shtml).Citation55
Phylogenetic analysis of select genes and whole genomes was performed using MEGA 5.1.0 (http://www.megasoftware.net/megamac.php), which accepts FASTA files for sequence alignment and uses a Maximum Composite Likelihood method that generated neighbor-joining and bootstrapped phylogenetic trees with 1000 bootstrap replications; for phylogenetic analysis; all other parameters were set by default.
Genome type identification
In silico restriction maps of available genome sequences from all HAdV-14 strains CHN/GZ01/2010, 303600, CHN/BJ430, CHN2012, and prototype de Wit were generated using the Vector NTI 10.3.0 software (Invitrogen Corp.; San Diego, CA, USA) as described in earlier studies.Citation56, Citation57, Citation58 These included profiles from restriction enzymes BamHI, BclI, Bgll, BglII, BstEII, EcoRI, HindIII, HpaI, Sall, SmaI, XbaI, and XhoI (TaKaRa Corp.; China), all of which were chosen in order to be consistent with the original nomenclature system devised by Li and colleagues.Citation59 Genome typing of these strains was determined by comparing its in silico RE profiles with other HAdV-14 genome types.Citation4, Citation34
GenBank accession numbers
The genome sequences used for phylogenic analyses are summarized in Table , with additional genome typing details given only for the type 14 strains (‘p’ denotes prototype; if no further designation is noted, the strain is the prototype). Sequences used for these studies included the E1A and hexon genes, either deposited as single gene entries in GenBank or which were extracted from the genome files.
Table 1 The genome, fiber, hexon, and E1A sequences of adenovirus species A–G used in this study
Results
Nucleotide sequence analysis of HAdV-B14p1 strain GZ01
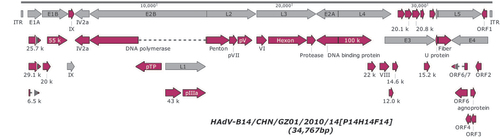
The genome of strain GZ01 was sequenced, assembled, annotated, and analyzed using computational methods. Figure presents the genomic organization and transcription map of GZ01. A total of 38 coding sequences were identified. These genome data were deposited in GenBank (accession number JQ824845) under the formal name, preferred by the National Center for Biotechnology Information (NCBI),Citation24 ‘Human adenovirus 14 isolate HAdV-B/CHN/GZ01/2010/14[P14H14F14]’; in this report, it is shortened to ‘‘GZ01’’. The genome comprises 34 767 bp, with a GC content of 48.83% that is consistent with the other members of subspecies B2 (mean of 49%).Citation60 A contemporaneous isolate BJ430 contains 34 762 bp.Citation32 These lengths are very similar to that of other HAdV-B14p1 strains 303600 (Lackland Air Force Base, USA; 2007) and CHN2012 (Beijing, China; 2012),Citation33 as well as the prototype from 1955; Genome sizes of 34 763 bp, 34 760 bp, and 34 764 bp, respectively.
Figure 1 Genomic organization and transcription map of HAdV-B14p1 strain GZ01. The grey arrows indicate the early, intermediate, and late transcription units; the red arrows indicate coding regions. Arrows reflect the direction of the coding transcripts.
Genome type of strains GZ01, BJ430, and CHN2012 as HAdV-B14p1
Genome type, based on restriction enzyme analysis (REA), was useful in the past for characterizing strains in the absence of full genome sequence data.Citation59, Citation61 It has limited use in the era of whole genome data, but may be useful in comparisons with strains reported in the literature with REA maps but are no longer available for futher analysis. The genome types of strains GZ01, BJ430, and CHN2012 were determined by in silico REA using the Vector NTI 10.3.0 software (Invitrogen Corp.; San Diego, CA, USA) and in comparison with the other HAdV-B14 strains Citation4, Citation34 (Figure). The REA profiles of GZ01, BJ430, and CHN2012 were consistent with and indistinguishable from that of HAdV-14p1 strain 303600, all which were confirmed to be genome type 14p1 according to the nomenclature published previously.Citation4, Citation34 It is clear, however, that genome type analysis by REA patterns may be misleading or incomplete as all the three China isolates are highly divergent from HAdV-14p1, as demonstrated by the indel mutations analysis. Furthermore, to support the observation that genome type identification may be misleading, it should be noted that while the initial wet-bench REA data suggested 303600 was a novel HAdV-B14a strain, it was disproven by the whole genome analysis.Citation4, Citation34
Figure 2 Identification of the genome types of HAdV-B14 strains GZ01 (Guangzhou), BJ430 (Beijing), and CHN2012 (Beijing). The in silico restriction enzyme analysis (REA) digestion patterns of HAdV-14 genomes were generated using the Vector NTI 10.3.0 software (Invitrogen Corp., San Diego, CA, USA). Genome digestion patterns of the strains are as follows: (A) de Wit or prototype (AY803294); (B) GZ01 (JQ824845); (C) 303600, reported as HAdV-B14p1 (FJ822614); (D) BJ430 (JN032132); and (E) CHN2012 (JX892927). These were analyzed with BamHI, BclI, Bgll, BglII, BstEII, EcoRI, HindIII, HpaI, SalI, SmaI, XbaI, and XhoI, as described by Li et al.Citation59 All the three recent China outbreak genomes correspond to the genome type of HAdV-14p1 and have identical REA profiles to each other. They are divergent from the HAdV-B14 prototype (1955). M: 1 kb DNA Sizing Ladder.

Inverted terminal repeat (ITR)
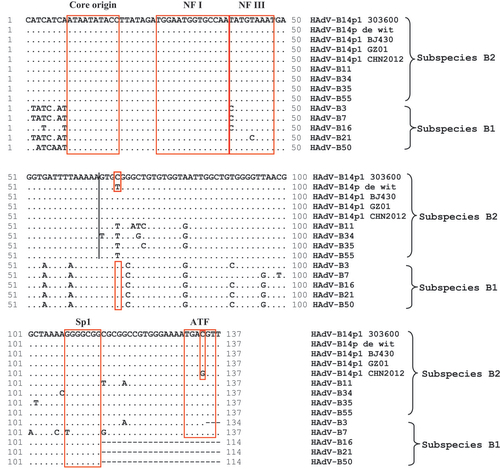
ITRs contain sequences encoding critical genome replication functions. The HAdV-B14p1 GZ01 genome has a 137 bp ITR that is identical to the ITRs of the other HAdV-B14p1 strains (BJ430, CHN2012, and 303600), except for a C to G mutation at nt 134 of strain CHN2012 (Figure). It is nearly identical to the ITRs reported for the other subspecies B2 members. The first 64 bases are completely identical amongst the prototype subspecies B2 ITRs, which is different for divergent subspecies B1. The binding sites for the host transcription factors NF I and NF III were identified in the ITR.Citation62, Citation63 These sequences have roles in enhancing virus replication and are necessary for efficient virus growth.Citation62, Citation63
Transcriptionally-related Sp1 and ATF binding sites were also conserved in subspecies B2 genomes. All four HAdV-B14p1 genomes differ from HAdV-B14p at position 68, with a C present, a transition mutation from the original T; this is also found at the complementary 3'-end, a validation that it is not a sequencing error. This particular T is conserved across all of the subspecies B2 prototypes, including HAdV-B55, but not amongst all the prototypes of the other subspecies, that is, all subspecies B1 members contain a C at this position; this is a marker to distinguish B1 from B2, and also HAdV-B14p1 from 14p. The nucleotide varies for the other six HAdV species (data not shown).
Figure 3 HAdV-B Inverted terminal repeats (ITR) analysis. An alignment of the left ITRs of select HAdV-B prototypes is presented, with two sets of critical sequence motifs (boxed): (1) Replication- Core origin, NF I and NF III binding sites, and (2) Sp1 and ATF binding sites. Nota bene, ‘.’ represent identical bases and ‘-’ represent deletions or gapping for sequence alignment. HAdV-B1: HAdV-B3, -B7, -B16, -B21, and -B50; HAdV-B2: HAdV-B11, -B14, -B34, -B35, and -B55.
Phylogenetic analysis of select genes and the whole genomes
Of the five recent China strains, only three were reported with whole genome data (GZ01, BJ430, and CHN2012). Phylogenetic analysis of the HAdV-B14 E1A, fiber, and hexon genes shows that the recent HAdV-B14p1 strains (strains GZ01, BJ430, CHN2012, 303600, 2971, and Dublin2009) are closely related to each other as well to the prototype and earlier reported HAdV-B14p genomes (1955 and 1974), as shown in Figure. The phylogenetic analysis of the whole genome sequences also confirms the sequence similarity with the prototype genome after approximately 50 years and across large geographical distances (Figure).
Figure 4 Phylogenetic analysis of the E1A, fiber, and hexon genes, as well as the whole genome sequences, from all HAdV-B14 strains and representatives of other HAdV species. Nucleotide sequences of the E1A, fiber, and hexon genes, as well as limited whole genomes, are available from GenBank. Taxon names include GenBank acc. no., isolation country, strain name, and year of isolation. Phylogenetic trees were generated by using the neighbor-joining method with 1000 replicates and constructed by the MEGA 5.1.0 software (http://www.ebi.ac.uk/tools/mafft). In these analyses, default parameters were applied, with a maximum-composite-likelihood model. Bootstrap numbers shown at the nodes indicate the percentages of 1000 replications producing the clade, with values above 80 considered robust. The scale bar is in units of nucleotide substitutions per site. The China HAdV-B14 strains were indicated with filled triangles.

Distribution of indel mutations across the genomes
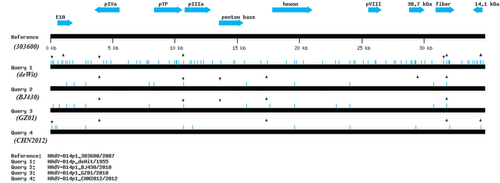
A global visualization of the alignment of genome sequences of GZ01, BJ430, 303600, CHN2012, and de Wit prototype is presented in Figure . This was generated using a ‘beta test version’ software, Genome Mutation Mapper (GMM; unpublished) that notes SNPs and indels of query genomes relative to a reference, and confirmed using DNA Sequencher (GeneCodes, Inc.; Ann Arbor, MI). The sequence similarities amongst them indicates that although these HAdV-B14p1 viruses have a common ancestor (type 14 serotype), their insertion/deletion (indel) patterns suggest two lineages. Indels and their subsequently inherited conserved patterns across genomes are key markers in determining lineages and for following molecular evolution, as reversion is highly unlikely.Citation64 Both strains GZ01 and BJ430 are highly divergent from the U.S. strain 303600, with respect to the indel patterns; there is one indel distinguishing between GZ01 and BJ430, a deletion at nt 29 481 in the BJ430 genome, that could be interpreted as GZ01 being more similar to 303600. CHN2012 contains a divergent indel pattern with respect to the other two China isolates. In addition to three deletions in common, it contains two additional indels (one each, insertion and deletion), relative to GZ01 and BJ430, that are uniquely also in the prototype genome. This may be indicative of two sub-lineages and independent introductions into China from abroad, or subsequent divergence from one. The indel numbers and patterns suggest there were at least two prototypes circulating in 1950s, time of the first prototype isolate, with one leading to 303600 and the second leading to the cluster of recent China strains. This visualization of the mutations alignment is critical to the understanding of the molecular evolution and lineages of pathogens as the low resolution genome percent nucleotide identities and REA (Figure) present very subtle and limited differences. GZ01, BJ430, CHN2012, and 303600 have highly similar percent identities in comparison with the prototype de Wit at 99.646%, 99.666%, 99.684%, and 99.689%, respectively.
Figure 5 Global genome mutation map visualization of the alignment of genomes from HAdV-14 strains de Wit, 303600, BJ430, GZ01, and CHN2012. To assess the lineages and molecular evolution relationships of these pathogens, Genome Mutation Mapper (GMM; unpublished beta version) is used to provide a global visualization of the nucleotide differences, including insertions and deletions (indels). The contemporaneously circulating and independently isolated HAdV-B14p1 strains have divergent, yet similar mutation profiles, with the indels defining their lineages. For this analysis, the genome of HAdV-14p1_303600/USA/2007 is set as the reference to which the other genomes are compared. Query 1: HAdV-B14p_deWit/The Netherlands/1955 (prototype); Query 2: HAdV-B14p1_BJ430/China//2010; Query 3: HAdV-14p1_GZ01/China/2010; and Query 4: HAdV-B14p1_CHN2012/China/2012. The triangles and inverted triangles indicate deletions and insertions, respectively; blue hash lines indicate base substitutions. Select gene markers are displayed for reference (locations are approximate), as are the genome size markers noted along the bottom.
Base substitution analysis of type 14 strains GZ01, BJ430, CHN2012, 303600, and de Wit
To complement and to check the global genome mutation map of the alignments, each point mutation amongst the five genomes are noted and compared, using the 2007 U.S. strain 303600 as reference (Table ). Base substitutions are indicative of recent and/or essential mutations as reversion may occur, unlike indel mutations.Citation64 The whole genome sequence of strain GZ01 is very similar to that of strain BJ430 with respect to mutations. Six mutation differences are noted between these genomes. These SNPs include two non-synonymous substitutions in the DNA polymerase (K to N) and protein VI genes (R to K). Four synonymous substitutions are noted for the E1A 29.1 K and 25.7 K genes, the L3 VI gene, and the E2A DNA binding protein gene. One mutation is in a non-coding region (nt 225) and two deletions, three T (nt 10 658) and one A (nt 13 266), in poly (T) and poly (A) regions, are also noted (Table ). Among these mutations, a deletion of A at nt 29 481 in the BJ430 genome distinguishes it from GZ01 (Figure ).
Table 2 Comparison of Mutations Across the Genomes of four HAdV-B14 Strains
When compared with strain 303600, 16 base substitutions were found in 11 coding regions of GZ01, which resulted in three non-synonymous substitutions in the E1A 6.5 K, E1B 54.9 K, and Protein VI coding regions, respectively. All of the indels occurred in non-coding regions, which led to length changes of the corresponding poly(A) or poly(T) sequences. Compared with the prototype de Wit strain, there were more mutations in strain GZ01, as would be expected given the time differences between their isolations: 93 base substitutions in the 31 genes, resulting in 35 non-synonymous substitutions. Although there were nine indels involving 25 nucleotides, only two of these resulted in non-synonymous substitutions: One in E1 29.1 K and 25.7 K coding regions (SV to I), with the other resulting the insertion of two amino acids KE in fiber knob domain, which is the most notable sequence difference between the HAdV-14p de Wit and all the other HAdV-14p1 strains (Table ). This was believed to have the potential of altering cell receptor binding and tissue tropisms, and hence pathogenicity, as the fiber knob recognizes the host cell receptor.Citation41
When compared with strain 303600, 11 base substitutions were found in strain CHN2012, which resulted in two non-synonymous substitutions in the E1B 54.9 K, E3 20.8 K coding regions, respectively. There are four synonymous substitutions in the E2B/L1 43 K, hexon, L4 100 K coding regions, respectively. The other mutations located at NCR, including one C to G mutation in ITR.
Discussion
China’s large, dense, and generally inaccessible population represents a unique environment for studying viral pathogens once they enter the population. The emergence, re-emergence, and transmission of a particular pathogen may be followed using high-resolution genome sequencing and, through computational methods, the molecular evolution and epidemiology of the pathogen may be revealed in great detail. According to the genome analysis of the three HAdV-B14 strains circulating in China, all three strains appear to be of separate, but related, lineages and may likely have been transmitted from abroad. Unlike the past, Beijing and Guangzhou are now ‘open’ to unrestrictive global travel and this may situate them to be the foci for the introduction of infectious disease agents to and from both overseas and other China provinces. Uniquely, Guangzhou has four additional direct long-standing connections to the international community. First, since the mid-1800s, many China emigrants to overseas destinations have originated from this region; therefore, there has ‘always’ been direct physical contact through homecoming visits. Second, Guangzhou has hosted several international events recently, bringing in an influx of visitors. For example, the Asian Games (Nov. 2010) was hosted in Guangzhou, bringing in more than 14 000 athletes along with a large number of foreign tourists and regional transient workers. Third, residents of other China provinces migrate to this prosperous region. Finally, Guangzhou is only 119 km from Hong Kong with the populations commingling; both populations are also in close contact with the global community, including Americans from both hemispheres, neighboring Asians, and Europeans, all of which have experienced respiratory diseases linked to the subspecies B2 HAdVs as well as other respiratory pathogens. For instance, from 2002 to 2003, the severe acute respiratory syndrome coronavirus (SARS-CoV) spread quickly from Guangzhou to Hong Kong and Beijing, and then across the country and globally, with high morbidity and mortality rates. Guangzhou, therefore, serves as an important focal point for infectious disease pathogen introduction, and surveillance is important in order to limit the public health impact on its population and elsewhere in China.
HAdV are important pathogens that appear to re-emerge after lengthy absences, either due to non-reporting because of diminished pathogenicity and infectivity or to perhaps latency or other mechanisms of cryptic infection. HAdV-B14, for example, re-emerged after approximately fifty years and HAdV-B7d re-emerged after twenty-one years.Citation56 The 2005 re-emergent HAdV-B14p1 strain was associated with several highly contagious and geographically wide-spread outbreaks of ARD, and included unexpected higher rates of fatalities.Citation11, Citation47, Citation65 These outbreaks occurred in both civilian (24 communities) and military (nine communities) populations in the United States (2005–2009) and Europe (2009–2011).Citation4, Citation5, Citation11, Citation47, Citation50, Citation65 Parenthetically and retrospectively, the first case identified was in a child in California (2003)Citation4 and was presumably the originating point for the subsequent larger military base outbreaks.Citation65
In a limited retrospective survey of respiratory infectious disease agents from patients at a hospital in Guangzhou (2010 and 2011), HAdVs were found and characterized with respect to types.Citation54 One of these isolates, typed, deposited, and noted in GenBank as an emergent and previously unreported HAdV type 14 in China (‘human adenovirus 14 isolate HAdV-B/CHN/GZ01/2010/14[P14H14F14’), was isolated from a throat swab of a 17-month-old child with acute suppurative tonsillitis (October 2010). Another simultaneously emergent HAdV-B14 strain BJ430 was isolated from a six-month-old infant diagnosed with bronchial pneumonia and hospitalized at the Beijing Children’s Hospital (December 2010). This is unusual because this condition had not been previously described for infections attributed to either HAdV-B14 or HAdV-B55, with its similar genome and disease symptoms.Citation11, Citation14, Citation48, Citation66 As HAdV-B14 has re-emerged globally recently, and since it is known as a highly contagious pathogen that has been associated with high hospitalization rates and fatality rates,Citation11, Citation47, Citation65 the emergence of this virus should set off an alarm that the proper surveillance of this virus is critical for large dense populations with naive immunity to HAdV-B14.
Comparative genomic analysis is leading to discovery of large numbers of novel molecular markers that are proving very helpful in understanding many important aspects of microbial phylogeny;Citation67, Citation68 of these molecular markers, the conserved indel mutations provide particularly useful means for identifying different groups of microbes in clear molecular terms and for understanding when they have branched off from a common ancestor.Citation67, Citation69, Citation70, Citation71 As reported here, indel mutations analysis suggests two co-circulating lineages from at least two co-circulating ‘prototype’ strains in 1950s, one giving rise to the 303600 strain and the other giving rise to the China cluster of HAdV-B14. Before the genomics era, many important adenovirus strains were characterized by REA. REA is still useful for characterizing current isolates by providing a bridge between their accessible genome data and the REA data that are only available in the literature, as important references and as historical isolates are no longer available for genomic analysis.Citation72 This report emphasizes that REA data should be carefully considered, as it may be misleading and incorrect. Although the REA analysis confirmed strains GZ01, BJ430, and CHN2012 as belonging to the same genome type 14p1 as strain 303600, high resolution analysis of the genome sequences, including indels, indicate different lineages and introductions into China, along with the absence of fatalities associated with 303600. Again, based on the genome mutation patterns, especially the indels, the de Wit prototype appears to be very different from the 303600 and is similar to the BJ430, GZ01, and CHN2012 strains. These may represent several lineages co-circulating ‘prototype 14’ strains, with one reported in the original study and the rest accumulating characteristic indel patterns that have been reported in this study. Most of the indels occur in the poly(A) and poly(T) regions, which have been proposed as high-resolution molecular strain markers for characterizing HAdV-14p1.Citation71 The recent report that HAdV-B14 dispersed from U.S. to Asia in 2007 via military traineesCitation53 provides another clue that the emergent HAdV-B14p1 strains in China may have been transmitted from abroad. However, as shown in this report, the whole genome data of that strain is critical for the correct interpretation of that possible transmission pattern.
The whole-genome sequences of strains GZ01 and BJ430 are nearly identical with each other, with respect to point mutations. One indel separates them. Strain GZ01 caused acute suppurative tonsillitis, an upper respiratory disease; however, strain BJ430 was associated with bronchial pneumonia, a lower respiratory tract disease. The reason for this difference in pathogenicity may be due to the genomic differences. A major difference between the two genomes is the two non-synonymous substitutions in the DNA polymerase (K to N) and protein VI genes (R to K), respectively (Table ). Of these two proteins, the 22-kDa cement protein VI is located beneath the peripentonal hexons in the viral capsid. It is identified as an endosomal membrane lytic factor, which is important for adenoviruses to overcome the barrier of the host cell membrane.Citation73 The non-synonymous substitution of R to K in the protein VI may lead to the function of protein VI being enhanced or weakened, which can presumably further change the HAdV tissue tropism. This is awaiting wet-bench studies.
Given the current ease of travel and global interactions, these ARD outbreaks associated with HAdV-B14 provide insights into the distribution, lineage, and molecular evolution of this pathogen. There had been no reports of HAdV-B14 in China prior to and during the period noted for HAdV-B14 re-emergence and circulation (2005–2009). This report presents the bioinformatics analysis of both HAdV-B14p1 strains in high resolution and detail, and provides putative lineages for these two surprisingly and intriguingly divergent, but, highly similar genomes linking Guangzhou and Beijing (2294 kilometers apart). A comparison with the re-emergent strain isolated in the U.S. (2007) as well as with the less similar but still remarkably conserved genome of the prototype virus from The Netherlands (1955) is also presented. The presentation of high resolution nucleotide sequence data and a map of mutations, particularly of the indels, provide detailed insight into two contemporaneously circulating human pathogens with divergent and parallel lineages. Both are noted as genome type HAdV-B14p1 via their low resolution but conveniently available REA patterns. The computational data presented in this report again demonstrates highly conserved but divergent HAdV-B14 genomes, which supports further studies of the epidemiology and molecular evolution of these related pathogens, and which, in turn, will provide a foundation for developing effective vaccines and public health strategies, including nationwide surveillance.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (31370199 and 31570155) and the Excellent Young Teacher Training Plan of Guangdong Province (Yq2013039), as well as by the Young Top-notch Talents of Guangdong Province Special Support Program (2014), and the Guangzhou Healthcare Collaborative Innovation Major Project (201400000002). Portions of this manuscript were completed at the Department of Ophthalmology, Howe Laboratory, Massachusetts Eye and Ear Infirmary, Harvard Medical School (Boston, Massachusetts, USA) as Q.Z. was funded by the China Scholarship Council (CSC No. 201508440056) as a Visiting Scholar (2015–2016); he thanks James Chodosh and Jaya Rajaiya for providing a stimulating intellectual environment. This project was additionally supported by a summer research grant (2016) to D.S. from the Office of the Vice President for Research at George Mason University. The Funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Related Research Data
References
- Pasarica M, Loiler S, Dhurandhar NV.Acute effect of infection by adipogenic human adenovirus Ad36. Arch Virol 2008;153: 2097–2102.
- Van Der Veen J, Kok G.Isolation and typing of adenoviruses recovered from military recruits with acute respiratory disease in The Netherlands. Am J Hyg 1957;65: 119–129.
- Kendall EJ, Riddle RW, Tuck HAet al.Pharyngo-conjunctival fever; school outbreaks in England during the summer of 1955 associated with adenovirus types 3, 7, and 14. Br Med J 1957;2: 131–136.
- Kajon AE, Lu X, Erdman DDet al.Molecular epidemiology and brief history of emerging adenovirus 14-associated respiratory disease in the United States. J Infect Dis 2010;202: 93–103.
- O'Flanagan D, O'Donnell J, Domegan Let al.First reported cases of human adenovirus serotype 14p1 infection, Ireland, October 2009 to July 2010. Euro Surveill 2011;16: pii=19801.
- Hierholzer JC, Pumarola A.Antigenic characterization of intermediate adenovirus 14-11 strains associated with upper respiratory illness in a military camp. Infect Immun 1976;13: 354–359.
- Chmielewicz B, Benzler J, Pauli Get al.Respiratory disease caused by a species B2 adenovirus in a military camp in Turkey. J Med Virol 2005;77: 232–237.
- Zhu Z, Zhang Y, Xu Set al.Outbreak of acute respiratory disease in China caused by B2 species of adenovirus type 11. J Clin Microbiol 2009;47: 697–703.
- Yang Z, Zhu Z, Tang Let al.Genomic analyses of recombinant adenovirus type 11a in China. J Clin Microbiol 2009;47: 3082–3090.
- Kajon AE, Dickson LM, Metzgar Det al.Outbreak of febrile respiratory illness associated with adenovirus 11a infection in a Singapore military training cAMP. J Clin Microbiol 2010;48: 1438–1441.
- Carr MJ, Kajon AE, Lu Xet al.Deaths associated with human adenovirus-14p1 infections, Europe, 2009-2010. Emerg Infect Dis 2011;17: 1402–1408.
- Zhang Q, Seto D, Zhao Set al.Genome sequence of the first human adenovirus type 14 isolated in China. J Virol 2012;86: 7019–7020.
- Kajon AE, Mistchenko AS, Videla Cet al.Molecular epidemiology of adenovirus acute lower respiratory infections of children in the South Cone of South America (1991–1994). J Med Virol 1996;48: 151–156.
- Walsh MP, Seto J, Jones MSet al.Computational analysis identifies human adenovirus type 55 as a re-emergent acute respiratory disease pathogen. J Clin Microbiol 2010;48: 991–993.
- Takeuchi S, Itoh N, Uchio Eet al.Adenovirus strains of subgenus D associated with nosocomial infection as new etiological agents of epidemic keratoconjunctivitis in Japan. J Clin Microbiol 1999;37: 3392–3394.
- Louie JK, Kajon AE, Holodniy Met al.Severe pneumonia due to adenovirus serotype 14: a new respiratory threat? Clin Infect Dis 2008;46: 421–425.
- Echavarria M.Adenoviruses in immunocompromised hosts. Clin Microbiol Rev 2008;21: 704–715.
- Stalder H, Hierholzer JC, Oxman MN.New human adenovirus (candidate adenovirus type 35) causing fatal disseminated infection in a renal transplant recipient. J Clin Microbiol 1977;6: 257–265.
- Kajon AE, Wadell G.Molecular epidemiology of adenoviruses associated with acute lower respiratory disease of children in Buenos Aires, Argentina (1984-1988). J Med Virol 1992;36: 292–297.
- Kaneko H, Aoki K, Ohno Set al.Complete genome analysis of a novel intertypic recombinant human adenovirus causing epidemic keratoconjunctivitis in Japan. J Clin Microbiol 2011;49: 484–490.
- Kaneko H, Aoki K, Ishida Set al.Recombination analysis of intermediate human adenovirus type 53 in Japan by complete genome sequence. J Gen Virol 2011;92 (Pt 6): 1251–1259.
- Walsh MP, Chintakuntlawar A, Robinson CMet al.Evidence of molecular evolution driven by recombination events influencing tropism in a novel human adenovirus that causes epidemic keratoconjunctivitis. PLoS ONE 2009;4: e5635.
- Robinson CM, Rajaiya J, Walsh MPet al.Computational analysis of human adenovirus type 22 provides evidence for recombination between human adenoviruses species D in the penton base gene. J Virol 2009;83: 8980–8985.
- Seto D, Chodosh J, Brister JRet al.Using the Whole-Genome Sequence To Characterize and Name Human Adenoviruses. J Virol 2011;85: 5701–5702.
- Liu EB, Ferreyra L, Fischer SLet al.Genetic analysis of a novel human adenovirus with a serologically unique hexon and a recombinant fiber gene. PLoS ONE 2011;6: e24491.
- Liu EB, Wadford DA, Seto Jet al.Computational and Serologic Analysis of Novel and Known Viruses in Species Human Adenovirus D in Which Serology and Genomics Do Not Correlate. PLoS ONE 2012;7: e33212.
- Robinson CM, Zhou X, Rajaiya Jet al.Predicting the next eye pathogen: analysis of a novel adenovirus. mBio 2013;4: e00595-12.
- Matsushima Y, Shimizu H, Kano Aet al.Novel human adenovirus strain, Bangladesh. Emerg Infect Dis 2012;18: 846–848.
- Zhou X, Robinson CM, Rajaiya Jet al.Analysis of human adenovirus type 19 associated with epidemic keratoconjunctivitis and its reclassification as adenovirus type 64. Invest Ophthalmol Vis Sci 2012;53: 2804–2811.
- Jones MS II, Harrach B, Ganac RDet al.New adenovirus species found in a patient presenting with gastroenteritis. J Virol 2007;81: 5978–5984.
- Hage E, Liebert UG, Bergs Set al.Human Adenovirus type 70: A novel, multiple recombinant species D adenovirus isolated from diarrheal faeces of a haematopoietic stem cell transplantation recipient. J Gen Virol 2015;96: 2734–2742.
- Tang L, An J, Xie Zet al.Genome and Bioinformatic Analysis of a HAdV-B14p1 Virus Isolated from a Baby with Pneumonia in Beijing. China. PLoS ONE 2013;8: e60345.
- Mi Z, Butt AM, An Xet al.Genomic analysis of HAdV-B14 isolate from the outbreak of febrile respiratory infection in China. Genomics 2013;102: 448–455.
- Houng HS, Gong H, Kajon Aet al.Genome sequences of human adenovirus 14 isolates from mild respiratory cases and a fatal pneumonia, isolated during 2006-2007 epidemics in North America. Respir Res 2010;11: 116.
- Lauer KP, Llorente I, Blair Eet al.Natural variation among human adenoviruses: genome sequence and annotation of human adenovirus serotype 1. J Gen Virol 2004;85 (Pt 9): 2615–2625.
- Dehghan S, Seto J, Liu EBet al.Computational analysis of four human adenovirus type 4 genomes reveals molecular evolution through two interspecies recombination events. Virology 2013;443: 197–207.
- Dehghan S, Seto J, Jones MSet al.Simian adenovirus type 35 has a recombinant genome comprising human and simian adenovirus sequences, which predicts its potential emergence as a human respiratory pathogen. Virology 2013;447: 265–273.
- Seto J, Walsh MP, Mahadevan Pet al.Genomic and bioinformatics analyses of HAdV-14p, reference strain of a re-emerging respiratory pathogen and analysis of B1/B2. Virus Res 2009;143: 94–105.
- Singh G, Robinson CM, Dehghan Set al.Overreliance on the Hexon Gene, Leading to Misclassification of Human Adenoviruses. J Virol 2012;86: 4693–4695.
- Purkayastha A, Ditty SE, Su Jet al.Genomic and bioinformatics analysis of HAdV-4, a human adenovirus causing acute respiratory disease: implications for gene therapy and vaccine vector development. J Virol 2005;79: 2559–2572.
- Wang H, Tuve S, Erdman DDet al.Receptor usage of a newly emergent adenovirus type 14. Virology 2009;387: 436–441.
- Singh G, Robinson CM, Dehghan Set al.Homologous Recombination in E3 Genes of Human Adenovirus Species D. J Virol 2013;87: 12481–12488.
- Lion T.Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev 2014;27: 441–462.
- Wadell G, Hammarskjold ML, Winberg Get al.Genetic variability of adenoviruses. Ann N Y Acad Sci 1980;354: 16–42.
- Wadell G.Molecular epidemiology of human adenoviruses. Curr Top Microbiol Immunol 1984;110: 191–220.
- De Jong JC, Wermenbol AG, Verweij-Uijterwaal MWet al.Adenoviruses from human immunodeficiency virus-infected individuals, including two strains that represent new candidate serotypes Ad50 and Ad51 of species B1 and D, respectively. J Clin Microbiol 1999;37: 3940–3945.
- Lewis PF, Schmidt MA, Lu Xet al.A community-based outbreak of severe respiratory illness caused by human adenovirus serotype 14. J Infect Dis 2009;199: 1427–1434.
- Prevention CfDCa.Acute respiratory disease associated with adenovirus serotype 14-four states, 2006-2007. MMWR Morb Mortal Wkly Rep 2007;56: 1181–1184.
- Girouard G, Garceau R, Thibault Let al.Adenovirus serotype 14 infection, New Brunswick, Canada, 2011. Emerg Infect Dis 2013;19: 119–122.
- Parcell BJ, McIntyre PG, Yirrell DLet al.Prison and community outbreak of severe respiratory infection due to adenovirus type 14p1 in Tayside, UK. Journal of Public Health 2014;37: 64–69.
- Huang G, Yu D, Zhu Zet al.Outbreak of febrile respiratory illness associated with human adenovirus type 14p1 in Gansu Province, China. Influenza Other Respi Viruses 2013;7: 1048–1054.
- Yu W, Sun H, Tian Jet al.Molecular epidemic characteristics analysis of an adenovirus infection outbreak. Chin J Public Health 2014;7: 972–974.
- Trei JS, Johns NM, Garner JLet al.Spread of adenovirus to geographically dispersed military installations, May-October 2007. Emerg Infect Dis 2010;16: 769–775.
- Han G, Niu H, Zhao Set al.Identification and typing of respiratory adenoviruses in Guangzhou, Southern China using a rapid and simple method. Virol Sin 2013;28: 103–108.
- Brudno M, Do CB, Cooper GMet al.LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res 2003;13: 721–731.
- Zhao S, Wan C, Ke Cet al.Re-emergent Human Adenovirus Genome Type 7d Caused an Acute Respiratory Disease Outbreak in Southern China After a Twenty-one Year Absence. Scientific reports 2014;4: 7365.
- Zhang Q, Su X, Seto Det al.Construction and characterization of a replication-competent human adenovirus type 3-based vector as a live-vaccine candidate and a viral delivery vector. Vaccine 2009;27: 1145–1153.
- Yu Z, Zeng Z, Zhang Jet al.Fatal community-acquired pneumonia in children caused by re-emergent human adenovirus 7d associated with higher severity of illness and fatality rate. Sci Rep 2016;6: 37216.
- Li QG, Zheng QJ, Liu YHet al.Molecular epidemiology of adenovirus types 3 and 7 isolated from children with pneumonia in Beijing. J Med Virol 1996;49: 170–177.
- Seto J, Walsh M, Mahadevan Pet al.Applying Genomic and Bioinformatic Resources to Human Adenovirus Genomes for Use in Vaccine Development and for Applications in Vector Development for Gene Delivery. Viruses 2010;2: 1–26.
- Li QG, Wadell G.Analysis of 15 different genome types of adenovirus type 7 isolated on five continents. J Virol 1986;60: 331–335.
- Mul YM, Verrijzer CP, van der Vliet PC.Transcription factors NFI and NFIII/oct-1 function independently, employing different mechanisms to enhance adenovirus DNA replication. J Virol 1990;64: 5510–5518.
- Hatfield L, Hearing P.The NFIII/OCT-1 binding site stimulates adenovirus DNA replication in vivo and is functionally redundant with adjacent sequences. J Virol 1993;67: 3931–3939.
- Nomura M, Benzer S.The nature of the ‘deletion’ mutants in the rII region of phage T4. J Mol Biol 1961;3: 684–692.
- Metzgar D, Osuna M, Kajon AEet al.Abrupt emergence of diverse species B adenoviruses at US military recruit training centers. J Infect Dis 2007;196: 1465–1473.
- Zhang Q, Seto D, Cao Bet al.Genome Sequence of Human Adenovirus Type 55, a Re-Emergent Acute Respiratory Disease Pathogen in China. J Virol 2012;86: 12441–12442.
- Gupta RSApplications of conserved indels for understanding microbial phylogeny. In:Oren A, Papke RT (eds). Molecular Phylogeny of Microorganisms.Norwich: Caister Academic Press.2010, 135–150.
- Naushad HS, Lee B, Gupta RS.Conserved signature indels and signature proteins as novel tools for understanding microbial phylogeny and systematics: identification of molecular signatures that are specific for the phytopathogenic genera Dickeya, Pectobacterium and Brenneria. Int J Syst Evol Microbiol 2014;64 (Pt 2): 366–383.
- Ajawatanawong P, Baldauf SL.Evolution of protein indels in plants, animals and fungi. BMC Evol Biol 2013;13: 140.
- Gao B, Gupta RS.Microbial systematics in the post-genomics era. Antonie Van Leeuwenhoek 2012;101: 45–54.
- Houng HS, Lott L, Gong Het al.Adenovirus microsatellite reveals dynamics of transmission during a recent epidemic of human adenovirus serotype 14 infection. J Clin Microbiol 2009;47: 2243–2248.
- Zhang Q, Dehghan S, Seto D.Pitfalls of restriction enzyme analysis in identifying, characterizing, typing, and naming viral pathogens in the era of whole genome data, as illustrated by HAdV type 55. Virol Sin 2016;31: 448–453.
- Wiethoff CM, Wodrich H, Gerace Let al.Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol 2005;79: 1992–2000.