
Abstract
Candida auris was first described in 2009, and it has since caused nosocomial outbreaks, invasive infections, and fungaemia across at least 19 countries on five continents. An outbreak of C. auris occurred in a specialized cardiothoracic London hospital between April 2015 and November 2016, which to date has been the largest outbreak in the UK, involving a total of 72 patients. To understand the genetic epidemiology of C. auris infection both within this hospital and within a global context, we sequenced the outbreak isolate genomes using Oxford Nanopore Technologies and Illumina platforms to detect antifungal resistance alleles and reannotate the C. auris genome. Phylogenomic analysis placed the UK outbreak in the India/Pakistan clade, demonstrating an Asian origin; the outbreak showed similar genetic diversity to that of the entire clade, and limited local spatiotemporal clustering was observed. One isolate displayed resistance to both echinocandins and 5-flucytosine; the former was associated with a serine to tyrosine amino acid substitution in the gene FKS1, and the latter was associated with a phenylalanine to isoleucine substitution in the gene FUR1. These mutations add to a growing body of research on multiple antifungal drug targets in this organism. Multiple differential episodic selection of antifungal resistant genotypes has occurred within a genetically heterogenous population across this outbreak, creating a resilient pathogen and making it difficult to define local-scale patterns of transmission and implement outbreak control measures.
A correction to this article is available online at https://doi.org/10.1038/s41426-018-0098-x.
Introduction
The emerging fungal pathogen Candida auris causes nosocomial invasive infections, predominantly in intensive care units (ICU). C. auris has a multidrug resistant (MDR) phenotypeCitation1, Citation2 with varying susceptibility to other azole drugs, amphotericin B and echinocandinsCitation3–Citation6 and acquired resistance to fluconazoleCitation7.
Since its first description in 2009 in JapanCitation8, C. auris infections have been reported in several countriesCitation1, Citation4, Citation6, Citation9–Citation16 with identification routinely carried out by matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS)Citation11, Citation14, Citation15. However, this method of identification can misidentify C. auris as other Candida species if commercial yeast identification databases are usedCitation17. Clonality has previously been identified within C. auris isolates from India, Brazil, South Africa, and South Korea using amplified fragment length polymorphism (AFLP) and multilocus sequence typing (MLST)Citation9, Citation14, Citation18. However, given the low discriminatory power and reproducibility of these techniques, genetic relatedness between isolates cannot be investigatedCitation14; while not currently routine in the investigation of fungal outbreaks, whole-genome sequencing (WGS) provides increased power to assess the relatedness of isolates to analyze patterns of nosocomial infections and global spreadCitation19.
In 2016, we described the first large-scale C. auris outbreak (April 2015–November 2016) occurring within a single specialist cardiothoracic hospital in LondonCitation11. Owing to the high uncertainty of the time and source of introduction of C. auris into the hospital, the rapid development of a molecular epidemiological toolkit was required. Outbreaks of other fungal pathogens have been previously investigated using short-read WGS, which provided sufficient information to discriminate between isolates and their phylogenetic relationships using single nucleotide polymorphism (SNP) analysisCitation20–Citation22. Recently, the handheld, portable MinION sequencer, manufactured by Oxford Nanopore Technologies, UK (ONT), has made rapid WGS widely available in the field and has been successfully used to analyze the molecular epidemiology of recent Ebola and Zika viruses outbreaksCitation23, Citation24.
Here, we describe the use of MinION nanopore sequencing technology on a pathogenic fungus to determine the genetic epidemiology of this fungal outbreak, both within the UK hospital and within a global context, alongside a reannotation of the genome of C. auris and definition of novel antifungal resistance alleles.
Results
We sequenced 25 clinical C. auris isolates from a recently described outbreakCitation11, which included eight isolates derived from four patients taken several days apart to establish possible within-patient diversity. In addition, we also sequenced two samples from the hospital environment to better represent the overall genetic diversity within the impacted hospital.
Rapid generation of outbreak C. auris reference genomes
We assembled five high-quality hybrid de novo reference genomes for C. auris using Illumina short-read sequences and MinION long-read sequences generated over 48 h. Five isolates were chosen to cover a range of dates (October 2015 to March 2016). Isolate 16B25 had the best overall assembly quality of 110 contigs, N50 = 396,317 bp and a total sequence length of 12.3 Mb (Table Supplementary Table S1). Approximately 98.94% of the 16B25 assembly mapped to the Pakistani C. auris genome B8441 assembled by Lockhart et al.Citation1 Further analysis revealed that the unmapped 1.06% of the 16B25 assembly was distributed throughout the genome and was likely attributed to genetic variation between the two isolates.
Summary of assembly and annotation statistics of the C. auris 16B25 genome, the C. auris B8441 referenceCitation1, the C. auris Ci 6684 referenceCitation26, and other pathogenic Candida species reference genomesCitation25
We generated an average of 5.2 million Illumina reads passing quality control for 27 isolates recovered during the outbreak that mapped closely (average 95.5%) to our reference genome (Supplemental Material Table S2). A total of 5366 protein coding genes, 4 rRNAs, and 156 tRNAs were predicted using the genome annotation pipeline described in Supplementary Methods. Table summarizes the general features of 16B25, along with those of other pathogenic Candida genomes. The number of protein coding genes presented here is in line with the predicted number of genes in C. lusitaniaeCitation25 (n = 5941), the closest known relative of C. auris.
There are fewer protein coding genes, tRNAs and rRNAs predicted in this genome than previously reported for C. auris Ci 6684 in Chatterjee et al.Citation26, as shown in Table . Running our annotation pipeline on the B8441 isolate presented in Lockhart et al.Citation1 found similar numbers of protein coding genes, rRNAs, and tRNAs (Table ). Therefore, the different total numbers between 16B25 and Ci 6684 are likely due to different annotation pipelines and not the quality of the reference assemblies. The number of protein coding genes identified in Chatterjee et al. (n = 8358) was substantially greater than that found in other Candida species (Table ) and is likely inflated due to over-prediction of short sequences, lack of filtering of repetitive sequences, and the use of only GenemarkS to predict the start of genes; our pipeline used additional criteria to achieve a predicted set of high-confidence genes.
Phylogenetic analysis reveals an Indian/Pakistani origin of C. auris outbreak
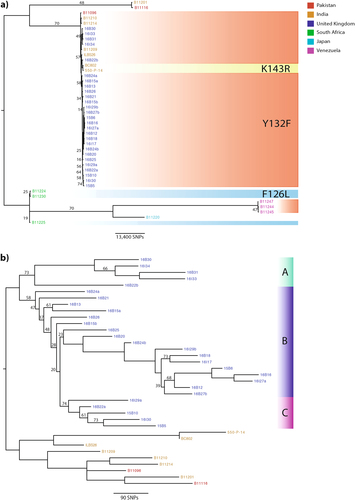
Phylogenetic analysis based on whole-genome SNPs revealed that the UK outbreak had an Indian/Pakistani origin (Fig. ). SNP calls for isolates from Venezuela, India, Pakistan, Japan, and South AfricaCitation1, Citation27 were also included to add geographic context to the outbreak. The UK outbreak isolates were in the same clade as those from India and Pakistan (Fig. ); on average, 240 SNPs separated UK outbreak isolates from isolates collected in India and Pakistan. There were no known patient travel links to India or Pakistan prior to admission into the hospital, however. We found an average of 103 SNPs separating isolates within the UK outbreak; later isolates in Clade A towards the end of the outbreak (Fig. ) exhibited 134 SNPs separating them compared to earlier isolates in Clades B and C isolated at the start of the outbreak (Fig. ), which showed an average of 90 SNPs separating them.
Branches were supported 75% or higher unless otherwise stated. Branch lengths represent the average expected rate of substitutions per site. a Outbreak isolates from the UK (shown in blue) were combined with isolates from around the globe, including India (orange), Pakistan (red), Venezuela (pink), Japan (turquoise), and South Africa (green), to infer a possible geographical origin. Isolates with known mutations in the ERG11 gene associated with resistance to fluconazole in C. albicans are shaded: Y132F in red, K143R in yellow, and F126L in blue. b Given the likely Indian/Pakistani origin of the outbreak isolates, phylogenetic analysis was repeated (as stated above), excluding isolates from South Africa, Venezuela, and Japan, to illustrate the UK outbreak. Isolates separating either into Cluster A (green), B (purple), or C (pink) are depicted to reflect likely introductions into the hospital
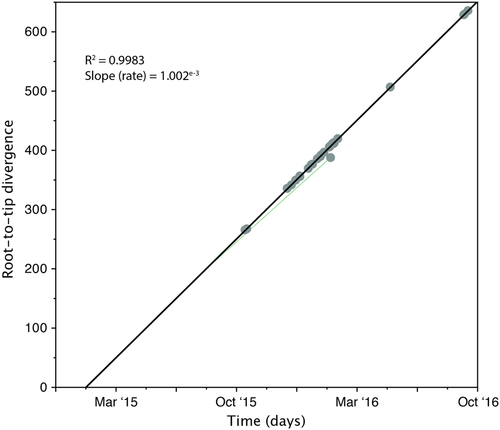
Fitting root-to-tip regression showed there was a linear relationship between sampling time (measured in days) and the expected number of nucleotide substitutions along the tree, demonstrating clock-like evolution across the timescale of the outbreak (Fig. ). There was a strong association between genetic distances and sampling dates (R2 = 0.9992), showing that the data set has a reasonable temporal signal for further molecular clock analysis. The evolutionary rate of nuclear DNA (calculated from the slope of the regression) equated to 1.002e−3 substitutions per generation, which is comparable to nuclear DNA of other fungal species, such as Schizosaccharomyces pombe beer strains (3.0e−3Citation28) and Saccharomyces cerevisiae (5.7e−3Citation29). The time to the most recent common ancestor (TMRCA) was estimated to be late January 2015, two months prior to the first patient identified with a C. auris infection. One isolate, 16B15b, showed less genetic divergence from the root than expected given its date of sampling, which was perhaps an indication of excessive passage or recombination.
Genetic distance is plotted against sampling time for the phylogeny of the C. auris outbreak. Each data point represents a tip on the phylogeny. The R2 for the regression and the slope, reflecting the evolutionary rate (in substitutions per site per day) is also shown
Clinical isolates of C. auris show multidrug resistance
Out of the 27 C. auris outbreak isolates, six displayed reduced susceptibility to two or more classes of antifungal drugs, and thirteen isolates displayed reduced susceptibility to two or more azole antifungal drugs (Table ). Only six isolates displayed reduced susceptibility to only one drug, fluconazole (Minimum Inhibitory Concentration (MIC): ≥256 µg/ml). Most (n = 24) isolates expressed elevated MICs to fluconazole (MIC: ≥256 µg/ml) with varying MICs to itraconazole (MIC: 0.03 µg/ml to ≥16 µg/ml), voriconazole (MIC: 0.12 µg/ml to 8 µg/ml) and posaconazole. Five isolates also displayed reduced susceptibility to amphotericin B (MIC: ≥2 µg/ml). Although resistance to amphotericin B is uncommon in Candida speciesCitation30, studies have associated mutations in the ERG2, ERG3, ERG5, ERG6, and ERG11 genes with the depletion of ergosterol and amphotericin B resistanceCitation30, Citation31. We did not identify any mutations in the aforementioned ERG genes in isolates displaying reduced susceptibility. Expanding the search across the genome found no candidate SNPs that were unique to isolates displaying reduced susceptibility.
MICs at time of isolation of all isolates included in this study and associated molecular resistance mechanism identified in this study
One isolate (16B15b) displayed elevated MICs to all echinocandin drugs (MIC: ≥8 µg/ml) but remained susceptible to all azole drugs, with the exception to fluconazole. 16B15b also displayed reduced susceptibility to flucytosine (MIC: ≥64 µg/ml), which was not seen in the other isolates; therefore, resistance and any associated mutations were unique to this outbreak isolate. This isolate belonged to a patient who received anidulafungin for 7 days for pancolitis, developing C. auris candidemia 11 days afterwards, at which point isolate 16B15a was recovered. Treatment was switched to amphotericin and 5-flucytosine for 2 weeks. 6 days after completing this treatment, pan-resistant C. auris (16B15b) was recovered from the vascular tip. Previous studies in Aspergillus and Candida species identified mutations in FKS1 associated with reduced echinocandin susceptibility or resistanceCitation32, Citation33; one non-synonymous SNP (nsSNP) causing a serine to tyrosine substitution (S652Y) was identified in the C. auris FKS1 gene from the 16B25 reannotated genome. Our updated annotation protocol comparing multiple lines of evidence predicted an extended first exon for FKS1 in comparison to the sequence submitted to GenBank (PIS58465); a comparison of the two protein sequences indicated that position 652 in the reannotated FKS1 sequence is analogous to position 639 in the GenBank entry, resulting in S639Y in FKS1 hotspot 1 (HS1). Another nsSNP caused a phenylalanine to isoleucine substitution (F211I) in the FUR1 gene identified in the 16B25 reannotated genome, which has a role in 5-flucytosine resistanceCitation34. Although neither of these mutations has been previously reported, single nucleotide changes in FUR1 in C. albicans and C. lusitaniae have been associated with 5-flucytosine resistanceCitation34, Citation35.
Orthologous sequences to C. albicans ERG11 were screened for substitutions that conferred known fluconazole resistance mutationsCitation36. The Y132F substitution in ERG11 was identified in all outbreak isolates, confirming an Indian/Pakistani origin. Lockhart et al.Citation1 also found that these substitutions were strongly correlated with geographic clades.
Interpretation of typing results in relation to epidemiology of the outbreak
C. auris outbreak isolates were grouped into three phylogenetic clusters (Clades A, B, and C; 18.5%, 63%, and 18.5%, respectively) (Fig. ). Clade A comprised five isolates from 2016 that had over 200 SNPs, distinguishing it from Clade B. On average, <100 SNPs separated the isolates in Clades B and C. Clade A was introduced into the hospital in early 2016 and formed the dominant outbreak strains towards the end of the outbreak; only 134 SNPs separated those isolates. Phenotypic antifungal susceptibility varied among Clade A isolates: all expressed elevated MICs to fluconazole (MIC: 128–256 µg/ml) and susceptibility to echinocandins (micafungin, caspofungin, and anidulafungin MIC: 0.06–0.12 µg/ml) and 5-flucytosine (MIC: 0.06 µg/ml) and two clinical isolates expressed reduced susceptibility to all azoles (MIC: 4–8 µg/ml posaconazole, 8 µg/ml voriconazole, 2–16 µg/ml itraconazole). This observation suggests that phenotypic antifungal susceptibility profiling cannot be reliably used to group genetically indistinguishable strains from nosocomial outbreaks.
Three patients with isolates in Clade A were admitted to the ICU. Two patients acquired C. auris while staying in the ICU, and another patient acquired C. auris within a surgical admission unit geographically placed next door to the high dependency unit in the same month, but this unit did not overlap with the ICU patients. Transmission of C. auris between different units within this hospital likely occurred via the movement of C. auris-positive patients or contaminated equipment. However, we are currently unable to establish routes of transmission in more detail because we did not sequence isolates from all patients during the outbreak and due to the heterogeneous nature of the founding population.
Epidemiology of individual patient transmission during the outbreak
Eight isolates within this study were sequential pairs of isolates from four separate patients (Table ); we hypothesized that there may have been nosocomial horizontal transmission between patients and/or their surrounding environment, as suggested in previous studiesCitation4, Citation9, Citation10, Citation18, for the following pairs of isolates: 16B22a and 16B22b from patient A (isolated 12 days apart); 16I27a (MICs were not carried out for this isolate) and 16B27b from patient B (isolated 1 day later); 16B24a and 16B24b from patient C (isolated 5 days apart); and 16B15a and 16B15b from patient D (isolated 32 days apart).
Clinical isolates of C. auris included in this study
In patient A, isolate 16B22a (recovered from the axilla) showed resistance to fluconazole only (Table ). The subsequent isolate from this patient, 16B22b (isolated from a central line tip), exhibited resistance to all azole drugs (Table ). Many SNPs (n = 277 SNPs) separated the two isolates (Fig. ) into distinct phylogenetic clusters (16B22a in Clade C and 16B22b in Clade A), suggesting independent acquisition of the infection by this patient within the unit. The two isolates from patient B similarly differed by many SNPs (n = 127 SNPs) but were both within Clade B. Given that 16I27a (from a body screen sample) was isolated 1 day prior to 16B27b (recovered from a positive blood culture), it is likely that this patient’s diversity represents an heterogenous C. auris population. In patient C, 16B24b was isolated from a clinical pacing wire sampled 5 days after the initial isolation of 16B24a from a sputum sample. These two isolates differ by 161 SNPs and were both placed in Clade B (Fig. ). In patient D, 16B15b showed raised MICs to all echinocandins and flucytosine, which were not seen in 16B15a. These two isolates were separated by 120 SNPs and were phylogenetically placed in Clade B (Fig. ). Our results suggest that none of these patients were infected with a single, clonally propagating C. auris strain; instead, the hospital (and all of its patients) was seeded with a genetically heterogenous population.
Three isolates (16B25, 16B20, and 16I29a) clustered closely together with an average difference of 99 SNPs. 16I29a was recovered from the environment around a C. auris-positive patient (the isolate was not sequenced) and was placed in Clade C (Fig. ). The patient from which 16B20 was recovered was present in an adjacent side room at the same time, suggesting a potential transmission between these patients, with 16I29a forming a new transmission chain of Clade C (Fig. ). 16B25 and 16B20 remained phylogenetically classified as Clade B (Fig. ). However, it remains unclear how the organism may have been transferred between the two rooms and patients. The third isolate of this cluster (16B25) was recovered from a patient present in the same room where 16I29a recovered. The patient was placed in this room 22 days after the previous C. auris-positive patient had left the room and became positive within 14 days of being in this isolation room.
Two isolates (16I29a and 16I29b) were isolated from the bed and trolley of a patient with a confirmed C. auris infection, who later died. However, MICs for these two environmental isolates were not carried out at the time of isolation. The genome sequence of the C. auris isolate from this patient was also not included in this study. A total of 161 SNPs differed between 16I29a and 16I29b, which were phylogenetically placed in Clades C and B, respectively. 16I29b appeared to be ancestral to a number of isolates in Clade B (Fig. ; 16B18, 16I17, 15B6, 16B16, 16I27a, 16B12, and 16B27b), which were, on average, separated by 122 SNPs.
Discussion
C. auris is an MDR fungal pathogen with intrinsically reduced susceptibility to antifungal drugsCitation37 that is capable of causing invasive infections. Here, we report WGS of C. auris infections from the largest UK outbreak to date, which occurred between April 2015 and November 2016 in a London hospital ICU and spread to two other wards.
A gold standard reference genome for the outbreak was assembled using long MinION-generated reads and Illumina short reads. While the GC nucleotide bias and base quality in >80% GC regions of the genome were similar in both Illumina and MinION sequencing, Illumina sequencing was more consistent across the whole genome (Figure S1). MinION reads displayed wide variation in base quality in >85% AT regions, ranging from a quality score of zero to 1.4, whereas Illumina reads ranged between quality scores of 0.75 and 0.85 for regions >85% AT. ONT has since released new chemistry that improves read quality and therefore variant calling, which may provide a competitive alternative to Illumina sequencing to further resolve transmission networks in outbreak settings and routine research.
Rapid generation of a genome assembly using MinION compliments the mapping of Illumina short reads to call high-confidence SNPs, which is of great importance in an outbreakCitation23, Citation24. Indeed, the use of Illumina data alone would not have yielded an assembly of similar quality (Table S1), highlighting the need for long reads, such as those generated by MinION. SNPs in ERG11 correlated with known C. albicans hotspotsCitation36, conferring resistance to the frontline drug fluconazole. Currently, no antifungal clinical breakpoints have been reported for C. aurisCitation38. All C. auris isolates in this study that exhibited high MICs to fluconazole (≥64 µg/ml) contained the Y132F amino acid substitution within ERG11, which is known to cause reduced susceptibility to fluconazoleCitation36, Citation37; seven isolates in which MIC determination was not carried out or the MIC was below the ECOFF of 64 µg/ml also contained amino acid substitutions known to cause reduced susceptibility to fluconazole. The ECOFFs at the time of publication are tentativeCitation3 and are not breakpoint MICs. Without performing the necessary allele swaps via gene deletion and observing wild-type phenotypes with lowered MICs to fluconazole, these ERG11 mutations cannot be confirmed as the sole mechanism of reduced susceptibility in C. auris. Thirteen isolates with reduced susceptibility to fluconazole also displayed elevated MICs for voriconazole and/or itraconazole. This cross-:“resistance” to multiple azole drugs has been reported in other studiesCitation4, Citation12, Citation39, yet no mechanism has been reported. Expression level changes of CDR1 have been associated with elevated itraconazole and fluconazole MICs in C. glabrataCitation40, whereas MDR1, MRR1, and GRP2 are differentially expressed in fluconazole and voriconazole resistant C. albicans isolatesCitation41. Differential expression of ERG11, ERG6, and UPC2 was also associated with posaconazole resistance in C. albicansCitation41. We did not identify a SNP-based mechanism responsible for the reduced posaconazole resistance in this study. Future studies therefore need to focus on the molecular biology of C. auris to elucidate the mechanisms of cross-resistance.
Five isolates displayed reduced susceptibility to amphotericin B, which although a rare event in Candida species, has been identified in up to 35% of C. auris isolates, suggesting an intrinsic resistance to this drugCitation1. No SNPs were associated with this reduced susceptibility, suggesting that a non-mutation-based mechanism of resistance is responsible. Thirteen isolates were MDR for more than one azole drug, and six displayed reduced susceptibility to two or more classes of antifungal drugs, posing an important clinical challenge in the treatment of these C. auris infections. Within this study, we have also highlighted reduced susceptibility to posaconazole in addition to reduced susceptibility to echinocandin and flucytosine in one MDR isolate.
Echinocandin resistance is linked to mutations in the FKS genes in other Candida speciesCitation42, and our analysis identified the S652Y mutation in FKS1 in an echinocandin-resistant isolate; because the annotation pipeline in this study predicted an extended first exon, this mutation is analogous to position 639 in the FKS1 sequence submitted to GenBank, resulting in a S639Y mutation in HS1. Previous studies have reported S639P and S639F mutations in C. auris FKS1Citation37, Citation43; this finding suggests that different mechanisms of echinocandin resistance are present within the C. auris population, which is similar to previous observations in ERG11 mutationsCitation1. Flucytosine resistance was observed in the same isolate and was associated with the F211I mutation in FUR1. Our analysis suggests that systemic echinocandin and flucytosine treatment can rapidly select for resistant genotypes across the outbreak timescale. Rapid MinION sequencing of C. auris isolates holds the promise of allowing the identification of drug resistance mutations, thus providing time-sensitive information in a clinical setting.
Phylogenomic analysis showed weak support for the monophyletic status of isolates within the hospital, suggesting that multiple introductions of C. auris occurred. On average, only 240 SNPs separated the UK outbreak isolates from the Indian/Pakistani clade, clearly showing that the UK outbreak had an Asian origin. When compared to other sequenced Asian C. auris isolates (Table S3) in Lockhart et al.Citation1, these SNP numbers suggest an anomalous amount of diversity within this outbreak. Future alignments will require clade-specific references due to the large evolutionary distances between the South American/African and Indian/Pakistani clades. Although the mode of introduction into the UK is unknown, temporal analysis of the outbreak isolates placed the most recent common ancestor as early March 2015, which correlates closely to the first confirmed infection within the hospital one month afterwards, suggesting a recent introduction into the UK.
Only 161 SNPs separated hospital environment isolates (bed and trolley of a confirmed C. auris-infected patient). C. auris was also recovered from inanimate surfaces, suggesting a population of genotypes are capable of contaminating the hospital environment, causing onward infection of human hostsCitation1. Given the SNP differences between isolates recovered from the same patient at different bodily locations (patient A, B, C, and D), the outbreak diversity is either due to multiple introductions, which is unlikely as all patients screened negative for C. auris upon admission, or a genetically heterogenous population seeded the hospital. Further, given the substantial number of SNPs separating the two isolates that were recovered from patient B 24 h apart, it is clear that genetically disparate isolates can infect the same host at the same time. The genomic diversity of C. auris within this outbreak makes mapping local-scale transmission events difficult as genetic bottlenecks may result in rapid changes in allele frequencies within local spatiotemporal scales. Clearly, sequencing multiple isolates of C. auris from patients alongside those from other UK outbreak settings is needed to more finely resolve the population genomic structure of this pathogen to better understand regional and local-scale transmission dynamics.
This study represents the first use of an ONT MinION sequencer on a human fungal pathogen; the rapid availability of long reads demonstrates that this technology is ideally used in an outbreak setting to provide high-quality contiguous assemblies. The association of a nsSNP in FUR1 with flucytosine resistance is both clinically relevant and novel. Although previous studies have reported FKS1 mutations, the S652Y mutation associated with echinocandin resistance presented here has not been previously reported. Further investigation into these mutations is required to confirm these associations.
Epidemiological analysis suggests that contact tracing was not sufficient for resolving fine-scale spatiotemporal processes across the outbreak due to multiple differential episodic selection events occurring across a genetically heterogenous C. auris population. Although the genomic approaches underpinning this study will likely be cornerstones of future research into this increasingly important pathogen, future research into C. auris should focus on sequencing many isolates from the same patient in multiple body sites to correctly establish the nature of persistence and transmission of C. auris within hospital environments. By combining molecular epidemiology with the traditional “shoe leather” epidemiology of contact tracing and interviewing patients, we will further our understanding of C. auris to achieve better control of future outbreaks.
Methods
Hospital and patient information
C. auris isolates were recovered from patients treated at the Royal Brompton Hospital (London, UK), a specialist cardiothoracic tertiary hospital. The outbreak was confined to adult patients in the mixed surgical and medical ICU, high dependency units (HDU) and the surgical admission ward (on a different floor). Details of the C. auris outbreak are reported in Schelenz et al.Citation11 Echinocandin treatment was prescribed in suspected C. auris infections. If a urinary tract infection was suspected, combination therapy of amphotericin B and 5-flucytosine was prescribed. Clinical data were collected as part of an approved Royal Brompton Hospital Trust service evaluation.
Fungal isolates
Twenty-seven C. auris isolates were studied, consisting of 25 clinical isolates from 21 patients and two isolates collected from the room of a patient known to be colonized with C. auris in the ICU (Table ). The identification of C. auris was conducted by matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometry (Bruker Daltonics, Fremont, CA, USA). An ethanol-formic acid extraction procedure was followed according to the manufacturer’s protocol for the identification of C. auris isolates. The spectra were analyzed using Flex Control 3.1 software (Bruker Daltonics Inc., Billerica, MA, USA) and MALDI Biotyper OC version 3.1 (Bruker Daltonics, Bremen, Germany). Scores were interpreted as >2.0 for species-level identification.
Storage of C. auris isolates
All Candida isolates were stored in 50% glycerol solution at −70 °C. Isolates were subcultured onto Sabouraud dextrose agar (SDA; Oxoid Ltd., Basingstoke, UK) and incubated at 35 °C (±2°C) in ambient air for 18−48 h to ensure adequate growth when required for further experimentation.
Antifungal susceptibility testing (AFST)
AFST of 9 systemic antifungal agents (amphotericin B, flucytosine, fluconazole, itraconazole, voriconazole, posaconazole, caspofungin, anidulafungin, and micafungin) was performed by colorimetric Sensititre YeastOne YST-10 broth dilution panels (Thermo Fisher Scientific, UK) according to the manufacturer’s instructions. Quality control was performed by testing Candida krusei ATCC® 6258 and Candida parapsilosis ATCC® 22019. All MICs were confirmed in the National Mycology Reference Laboratory, Public Health England, Bristol, UK, using broth microdilution according to CLSI method M27-A3Citation44.
gDNA extraction for MinION sequencing
C. auris isolates were grown in 10 ml of YPD broth (Sigma-Aldrich, UK) for 48 h at 37°C with agitation (180 rpm). High-molecular weight genomic DNA was extracted using the standard MasterPure Yeast DNA purification kit (Epicentre Biotechnologies, Cambridge, UK). Extracted gDNA was quantified using a Qubit 2.0 fluorometer and dsDNA BR (double-stranded DNA, broad range) assay kit (Life Technologies, Carlsbad, CA, USA). Quality control of gDNA prior to library preparation was performed using gDNA ScreenTape assays on the TapeStation 2200 system (Agilent, Santa Clara, CA, USA). Purified gDNAs were stored at 4°C until library preparation.
High-molecular-weight DNA (1 µg) was fragmented using a Covaris G-Tube (Covaris, Woburn, USA) at 4200 rpm. Fragmented DNA was end-repaired using the NEBNext Ultra II (New England Biolabs, Ipswich, USA), and cleaned using Agencourt AMPure XP beads (Beckman Coulter, USA) with a ratio of 1:1 beads to DNA mixture. End-repaired DNA was then A-tailed using the NEB Blunt/TA-ligase master mix (New England Biolabs, Ipswich, USA). The sequence-ready library was purified using MyOne Streptavidin C1 beads (Thermo Fisher Scientific, USA) and diluted prior to loading into the MinION flowcell. A 48-hour sequencing protocol was initiated using the MinION control software MinKNOW (v0.51.1.62). Read event data were base-called by the software Metrichor (v2.39.3). Each library was run on two flow cells to improve overall coverage.
gDNA extraction for Illumina sequencing
High-molecular weight genomic DNA was extracted with an optimized MasterPure Yeast DNA purification kit (Epicentre Biotechnologies, Cambridge, UK) with an additional bead-beating step. Extracted gDNA was quantified as mentioned previously. Purified gDNA was stored at 4 °C until library preparation. All Nextera library preparation and Illumina HiSeq 2500 sequencing of 2 × 250 bp paired end reads was carried out by MicrobesNG (University of Birmingham, UK). Four additional isolates (16B30, 16B31, 16I33, and 16I34) from a later stage of the outbreak (from June 2016 to October 2016) were sequenced using TruSeq Nano library preparation and Illumina HiSeq 2500 sequencing of 2 × 250 bp paired end reads at The Centre for Genomic Pathogen Surveillance (Wellcome Genome Campus, Cambridge, UK).
Library preparation and sequencing
Five clinical isolates (Table ) representing a 146-day time frame were chosen for sequencing using the handheld MinION sequencer (Oxford Nanopore Technologies, Oxford, UK) to generate whole genome references and provide genome annotations to observe the merit of the sequencing technology.
Twenty-three isolates (Table ), which include the five clinical isolates sequenced using MinION, were chosen for the current gold standard (Illumina sequencing) using the Nextera library preparation method as part of the MicrobesNG service (University of Birmingham, UK). The remaining four isolates were sequenced using TruSeq library preparation. These isolates represented a 155-day time frame and included isolates from patients and the environment around infected patients. Isolates of C. auris for both MinION and Illumina sequencing were cultured as described in supplementary methods. All raw reads in this study have been submitted to the European Nucleotide Archive under project accession PRJEB20230. Details of genome assembly and annotation are described in supplementary methods.
Assembly of MinION and Illumina reads
MinION reads were extracted using poReCitation45. Hybrid assembly of Illumina and nanopore reads was performed for each isolate using SPAdes (v3.9.0) with default k-mer lengths (21, 33, and 55) and 1D scaffolding. An assembly statistics summary for each hybrid assembly is provided in Table S1. The hybrid assembly of isolate 16B25 provided the best statistics, so it was used as a reference for SNP calling. The genome assembly has been submitted to NCBI under the project accession PRJNA392455.
Genome annotation
The C. auris genome was annotated using GenemarkCitation46, BLASTx against SwissProtCitation47 and KEGGCitation48, and HMMER hmmscanCitation49 against PFAMCitation50 and TIGRFAMCitation51. We ran tRNAscanCitation52 and RNAmmerCitation53 to identify non-protein coding genes. Gene predictions were checked for a variety of issues, including overlap with non-coding genes, overlap with coding genes, and the presence of in-frame stops. Genes were named according to BLAST and HMMER evidence in the following order of precedence: (1) SwissProt, (2) TIGRfam, and (3) KEGG, where BLAST hits must meet the 70% identity and 70% overlap criteria to be considered a good hit and for the name to be applied. Otherwise, genes were named hypothetical protein. The gene annotations can be found at https://figshare.com/articles/Candida_auris_gene_annotations/5203618.
Alignment of Illumina reads and phylogenetic analysis
Raw Illumina reads were quality checked using FastQC (v0.11.3; Babraham Institute) and aligned to the hybrid 16B25 reference genome using Burrows-Wheeler Aligner (BWA) v0.7.8 memCitation54 and converted to sorted BAM format using SAMtools v1.3.1Citation55. Picard v2.6.0 and GATK v3.6Citation56 were used to pre-process the alignments prior to variant calling. Base recalibration was also performed using GATK. Variants were called using GATK HaplotypeCaller, excluding repetitive regions as identified using RepeatMasker v4.0.6Citation57, and filtered using parameters “DP < 10||MQ < 40.0||QD < 2.0||FS > 60.0||ABhom < 0.9”. Next, all variant calls that had a less than a minimum genotype quality of 50 were removed.
Phylogenies for whole genome SNP data were constructed and visualized as described in Rhodes et al.Citation58 The rate of evolution (represented as the number of substitutions per day) along the tree topology was estimated using TempEst v1.5.1Citation59 and calibrated with sampling times. Root-to-tip regression was calculated, and the root of the tree was selected to maximize R2.
Benchmarking against CDC bioinformatics pipeline
Our bioinformatics pipeline was also used to call SNPs from isolates within the Indian/Pakistani clade presented in Lockhart et al.Citation1 to confirm the genetic diversity between UK C. auris isolates in the outbreak; the results are presented in Table S3.
Mutation identification in ERG11, FKS1, and FUR1 genes
Orthologous sequences to C. albicans ERG11 (SC5314) were extracted from each C. auris genome. Sequences were evaluated for amino acid substitutions to mutations within hot spot regions in C. albicansCitation36 as described in Lockhart et al.Citation1 Predicted FKS1 and FUR1 genes from the genome annotation were used to identify the presence or absence of mutations in C. auris isolates.
Supplementary information
Download MS Word (100.5 KB)Acknowledgements
We thank Oxford Nanopore Technologies for their generous contribution of flow cells and nanopore sequencing kits. We also wish to extend our thanks to Nicholas J. Loman (University of Birmingham) for providing insight into the initial conception of experiments and to Anastasia Litvintseva, Nancy A.M. Chow, and Kizee Etienne (Centers for Disease Control, USA) for providing additional sequence data. We also thank the sequencing team at Wellcome Trust Sanger Institute for sequencing four of the isolates presented here. J.R. was supported by an Antimicrobial Research Collaborative (ARC) early career research fellowship (RSRO_54990). R.A.F. was supported by an MIT/Wellcome Trust Fellowship. M.C.F. was supported by the Natural Environmental Research Council (NERC: NE/K014455/1) and the Medical Research Council (MRC: MR/K000373/1). D.A.-J. was supported by NERC and the Wellcome Trust. C.A.C was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Grant No. U19AI110818. D.M.A. supported by the Wellcome Trust (Grant No. 099202). Genome sequencing was provided by MicrobesNG (http://www.microbesng.uk), which is supported by the BBSRC (Grant No. BB/L024209/1).
Authors’ contributions
Clinical and outbreak data analysis: S.S. Collected isolates: S.S. Conceived experiments: J.R., M.C.F., D.A.-J., and S.S. DNA extractions: A.A., J.R. MinION DNA sequencing: J.R. Illumina sequencing: D.M.A. and MicrobesNG. Bioinformatic analysis: J.R. Genome annotation: R.A.F. and C.A.C. Manuscript preparation: J.R., R.A.F., A.A, and S.S.
Conflict of interest
J.R. received flow cells and reagents from Oxford Nanopore Technologies (ONT) free of charge, and has also presented work at a conference hosted by ONT. Authors report no other conflict of interest.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41426-018-0045-x).
Related Research Data
References
- LockhartSRSimultaneous emergence of multidrug resistant Candida auris on three continents confirmed by whole genome sequencing and epidemiological analysesClin. Infect. Dis.201764 134 14010.1093/cid/ciw691
- LamothFLockhartSRBerkowELCalandraTChanges in the epidemiological landscape of invasive candidiasisJ. Antimicrob. Chemother.201873i4i1310.1093/jac/dkx444
- ArendrupMCPrakashAMeletiadisJSharmaCChowdharyAComparison of EUCAST and CLSI reference microdilution MICs of eight antifungal compounds for Candida auris and associated tentative epidemiological cutoff valuesAntimicrob. Agents Chemother.201761e00485175444165
- Calvo, B. et al. First report of Candida auris in America: clinical and microbiological aspects of 18 episodes of candidemia. J. Infect.73, 369–374 (2016).
- SarmaSCandidemia caused by amphotericin B and fluconazole resistant Candida aurisIndian J. Med. Microbiol.201331909110.4103/0255-0857.108746
- MagoboRECorcoranCSeetharamSGovenderNPCandida auris-associated candidemia, South AfricaEmerg. Infect. Dis.2014201250125110.3201/eid2007.1317654073876
- LockhartSRBerkowELChowNWelshRMCandida auris for the clinical microbiology laboratory: not your grandfather’s Candida speciesClin. Microbiol. Newsl.2017399910310.1016/j.clinmicnews.2017.06.0035831145
- SatohKCandida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospitalMicrobiol. Immunol.200953414410.1111/j.1348-0421.2008.00083.x
- ChowdharyANew clonal strain of Candida auris, Delhi, IndiaEmerg. Infect. Dis.2013191670167310.3201/eid1910.1303933810747
- LeeWGFirst three reported cases of nosocomial fungemia caused by Candida aurisJ. Clin. Microbiol.2011493139314210.1128/JCM.00319-113165631
- Schelenz, S. et al. First hospital outbreak of the globally emerging Candida auris in a European hospital. Antimicrob. Resist. Infect. Control5, 35 (2016). 10.1186/s13756-016-0132-5
- Ben-AmiRMultidrug-resistant Candida haemulonii and C. auris, Tel Aviv, Israel.Emerg. Infect. Dis20172319520310.3201/eid2302.1614865324804
- SchwartzISHammondGWFirst reported case of multidrug-resistant Candida auris in CanadaCan. Commun. Dis. Report.20174315015310.14745/ccdr.v43i78a02
- PrakashAEvidence of genotypic diversity among Candida auris isolates by multilocus sequence typing, matrix-assisted laser desorption ionization time-of-flight mass spectrometry and amplified fragment length polymorphismClin. Microbiol. Infect.201622277.e1910.1016/j.cmi.2015.10.022
- MohsinJThe first cases of Candida auris candidaemia in OmanMycoses20176056957510.1111/myc.12647
- AraúzABIsolation of Candida auris from 9 patients in Central America: importance of accurate diagnosis and susceptibility testingMycoses201861444710.1111/myc.12709
- LockhartSRThinking beyond the common Candida species: need for species-level identification of Candida due to the emergence of multidrug-resistant Candida aurisJ. Clin. Microbiol.2017553324332710.1128/JCM.01355-175703798
- ChowdharyAMultidrug-resistant endemic clonal strain of Candida auris in IndiaEur. J. Clin. Microbiol. Infect. Dis.20133391992610.1007/s10096-013-2027-1
- AsadzadehMAhmadSAl-SweihNKhanZMolecular fingerprinting studies do not support intrahospital transmission of Candida albicans among candidemia patients in KuwaitFront. Microbiol.2017824710.3389/fmicb.2017.002475318450
- LitvintsevaAPWhole-genome analysis of Exserohilum rostratum from an outbreak of fungal meningitis and other infectionsJ. Clin. Microbiol.2014523216322210.1128/JCM.00936-144313140
- EtienneKAWhole genome sequence typing to investigate the Apophysomyces outbreak following a tornado in Joplin, Missouri, 2011PLoS ONE20127e4998910.1371/journal.pone.00499893507928
- EngelthalerDMNext-generation sequencing of Coccidioides immitis Isolated during cluster investigationEmerg. Infect. Dis.20111722723210.3201/eid1702.1006203204756
- Quick, J. et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol. 16, 114 (2015).
- Quick, J. et al. Real-time, portable genome sequencing for Ebola surveillance. Nature530, 228–232 (2016).
- ButlerGEvolution of pathogenicity and sexual reproduction in eight Candida genomesNature200945965766210.1038/nature080642834264
- ChatterjeeSDraft genome of a commonly misdiagnosed multidrug resistant pathogen Candida aurisBMC Genom.20151611610.1186/s12864-015-1863-z
- SharmaCKumarNPandeyRMeisJFChowdharyAWhole genome sequencing of emerging multidrug resistant Candida auris isolates in India demonstrates low genetic variationNew Microbes New Infect.201613778210.1016/j.nmni.2016.07.0035006800
- JeffaresDCThe genomic and phenotypic diversity of Schizosaccharomyces pombeNat. Genet.20154723524110.1038/ng.32154645456
- LitiGPopulation genomics of domestic and wild yeastsNature200945833734110.1038/nature077432659681
- Cuenca-EstrellaMAntifungal drug resistance mechanisms in pathogenic fungi: from bench to bedsideClin. Microbiol. Infect.201420545910.1111/1469-0691.12495
- ArendrupMCPattersonTFMultidrug-resistant candida: epidemiology, molecular mechanisms, and treatmentJ. Infect. Dis.2017216S445S45110.1093/infdis/jix131
- Jimenez-Ortigosa, C., Moore, C., Denning, D. W. & Perlin, D. S. Emergence of echinocandin resistance due to a point mutation in the fks1 gene of Aspergillus fumigatus in a patient with chronic pulmonary Aspergillosis. Antimicrob. Agents Chemother. AAC.01277–17–15. 10.1128/AAC.01277-17 (2017).
- Ben-AmiRFitness and virulence costs of Candida albicans FKS1 hot spot mutations associated with echinocandin resistanceJ. Infect. Dis.201120462663510.1093/infdis/jir3513144175
- DodgsonARDodgsonKJPujolCPfallerMASollDRClade-specific flucytosine resistance is due to a single nucleotide change in the FUR1 gene of Candida albicansAntimicrob. Agents Chemother.2004482223222710.1128/AAC.48.6.2223-2227.2004415630
- PaponNMolecular mechanism of flucytosine resistance in Candida lusitaniae: contribution of the FCY2, FCY1, and FUR1 genes to 5-fluorouracil and fluconazole cross-resistanceAntimicrob. Agents Chemother.20075136937110.1128/AAC.00824-06
- MorioFLogeCBesseBHennequinCLe PapePScreening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literatureDiagn. Microbiol. Infect. Dis.20106637338410.1016/j.diagmicrobio.2009.11.006
- ChowdharyAA multicentre study of antifungal susceptibility patterns among 350 Candida auris isolates (2009–17) in India: role of the ERG11 and FKS1 genes in azole and echinocandin resistanceJ. Antimicrob. Chemother.201813e10062909
- Jeffery-SmithACandida auris: a review of the literatureClin. Microbiol. Rev.201831e0002917
- Ruiz GaitánACNosocomial fungemia by Candida auris: first four reported cases in continental EuropeRev. Iberoam. Micol.201734232710.1016/j.riam.2016.11.002
- SanguinettiMMechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistanceAntimicrob. Agents Chemother.20054966867910.1128/AAC.49.2.668-679.2005547307
- WhaleySGAzole antifungal resistance in Candida albicans and emerging non-albicans Candida speciesFront. Microbiol.20177112210.3389/fmicb.2016.02173
- Garcia-EffronGLeeSParkSClearyJDPerlinDSEffect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-d-glucan synthase: implication for the existing susceptibility breakpointAntimicrob. Agents Chemother.2009533690369910.1128/AAC.00443-092737881
- Berkow, E. L. & Lockhart, S. R. Activity of CD101, a long-acting echinocandin, against clinical isolates of Candida auris. Diagn. Microbiol. Infect. Dis.10.1016/j.diagmicrobio.2017.10.021 (2017).
- Rex, J. H., Alexander, B. D., Andes, D. & Arthington-Skaggs, B. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts. Approved Standard. 3rd edn (M27-A3) (Clinical and Laboratory Standards Institute (CLSI), Wayne, PA, 2008).
- WatsonMpoRe: an R package for the visualization and analysis of nanopore sequencing dataBioinformatics20153111411510.1093/bioinformatics/btu590
- BorodovskyMMcIninchJGENMARK: parallel gene recognition for both DNA strandsComput. Chem.19931712313310.1016/0097-8485(93)85004-V
- BairochAApweilerRThe SWISS-PROT protein sequence database and its supplement TrEMBL in 2000Nucleic Acids Res.200028454810.1093/nar/28.1.4510592178
- KanehisaMGotoSKEGG: Kyoto encyclopedia of genes and genomesNucleic Acids Res.200028273010.1093/nar/28.1.27102409
- FinnRDClementsJEddySRHMMER web server: interactive sequence similarity searchingNucleic Acids Res.201139W29W3710.1093/nar/gkr3673125773
- FinnRDPfam: the protein families databaseNucleic Acids Res.201442D222D23010.1093/nar/gkt1223
- HaftDHSelengutJDWhiteOThe TIGRFAMs database of protein familiesNucleic Acids Res.20033137137310.1093/nar/gkg128165575
- LoweTMEddySRtRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequenceNucleic Acids Res.19972595596410.1093/nar/25.5.0955146525
- LagesenKRNAmmer: consistent and rapid annotation of ribosomal RNA genesNucleic Acids Res.2007353100310810.1093/nar/gkm1601888812
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM (2013). arXiv:1303.3997
- LiHThe sequence alignment/map format and SAMtoolsBioinformatics2009252078207910.1093/bioinformatics/btp3522723002
- McKennaAThe genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing dataGenome Res.2010201297130310.1101/gr.107524.11020644199
- Smit, A., Hubley, R., & Green, P. RepeatMasker Open-4.0. 2013–2015 (2015).
- RhodesJA population genomics approach to assessing the genetic basis of within-host microevolution underlying recurrent Cryptococcal meningitis infectionG3201771165117610.1534/g3.116.037499
- RambautALamTTMax CarvalhoLPybusOGExploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen)Virus Evol.20162vew00710.1093/ve/vew0074989882
- PfallerMADiekemaDJProgress in antifungal susceptibility testing of Candida spp. by use of clinical and laboratory standards institute broth microdilution methods, 2010 to 2012J. Clin. Microbiol.2012502846285610.1128/JCM.00937-123421803