
Abstract
Hand, foot, and mouth disease (HFMD) caused by enteroviruses remains a public health threat, particularly in the Asia-Pacific region during the past two decades. Moreover, the introduction of multiple subgenotypes and the emergence of recombinant viruses is of epidemiological importance. Based on either the full genome or VP1 sequences, 32 enteroviruses (30 from HFMD patients, 1 from an encephalitic patient, and 1 from an asymptomatic contact case) isolated in Thailand between 2006 and 2014 were identified as 25 enterovirus 71 (EV71) isolates (comprising 20 B5, 1 C2, 2 C4a, and 2 C4b subgenotypes) and 7 coxsackievirus A16 (CA16) isolates (comprising 6 B1a and 1 B1b subgenotypes). The EV71 subgenotype C4b was introduced into Thailand for the first time in 2006 and was replaced by subgenotype C4a strains in 2009. Phylogenetic, similarity plot and bootscan analyses of the complete viral genomes identified 12 recombinant viruses among the 32 viral isolates. Only one EV71-B5 isolate out of 20 was a recombinant virus with one region of intratypic or intertypic recombination, while all four EV71-C4 isolates were recombinant viruses having undergone double recombination, and all seven CA16 isolates were recombinant viruses. The recombination breakpoints of these recombinants are located solely within the P2 and P3 regions. Surveillance for circulating strains and subgenotype replacement are important with respect to molecular epidemiology and the selection of the upcoming EV71 vaccine. In addition, the clinical importance of recombinant viruses needs to be further explored.
Introduction
Hand, foot, and mouth disease (HFMD) is an acute febrile illness characterized by vesicular eruptions on the palms of the hands, the feet, and in the oral cavity and is common in children under 5 years of age worldwideCitation1–Citation4. HFMD was recognized as an emerging disease after large outbreaks of the disease in Malaysia in 1997 and Taiwan in 1998Citation5–Citation8. Subsequently, outbreaks of HFMD were reported in several part of the Asia-Pacific region. Currently, HFMD is an endemic disease in the People’s Republic of China, Taiwan, and several Southeast Asian countries, such as Vietnam and CambodiaCitation9–Citation15. In Thailand, HFMD has been listed as a disease to be notified to the Ministry of Public Health since 2001.
HFMD is caused by several enteroviruses, with the major etiologic agents being enterovirus 71 (EV71) and coxsackievirus A16 (CA16)Citation3,Citation16. Compared with CA16, EV71 is associated with relatively more severe symptoms and complications that may involve central nervous system and cardiopulmonary complications, such as brain stem encephalitis, autonomous nervous system dysregulation, neurological sequelae, neurogenic pulmonary edema, and myocarditisCitation1,Citation4,Citation17,Citation18. According to epidemiological data, neurological complications in EV71-associated HFMD have varied among outbreaks, suggesting that the viral genetic background has an effect on the disease presentationCitation11,Citation15.
EV71 and CA16 belong to the species Enterovirus A (EV-A), genus Enterovirus, family Picornaviridae. Members of this species have small, nonenveloped viral particles with icosahedral symmetry containing a positive sense, single-stranded RNA genome that is ~7.4 kilobases (kb) in lengthCitation19,Citation20. The genome is flanked by a 5ʹ-untranslated region (5ʹ-UTR) adjacent to the virus-encoded peptide (VPg) and a 3ʹ-untranslated region (3ʹ-UTR) adjacent to a poly(A) tail. The genome comprises one open reading frame (ORF) encoding a polyprotein that is divided into three parts, P1, P2, and P3. The P1 region encodes four structural viral proteins (VP1–VP4), while P2 encodes three nonstructural proteins (2A–2C) and P3 encodes for four nonstructural proteins (3A–3D)Citation3,Citation4,Citation21. VP1, the major antigenic and neutralizing domain, is the most variable protein. Based on VP1, EV71 is classified into three genotypes, namely, A, B, and C. Genotype A contains a single member, the BrCr prototype, which was initially identified in California, USA, in 1970 from a patient with encephalitisCitation21,Citation22. Genotypes B and C are further subdivided into five subgenotypes, including B1-B5 and C1-C5, respectivelyCitation4,Citation21. Similarly, based on VP1 sequences, CA16 is divided into two lineages (A and B). The prototype G-10 is the only member of lineage A, while lineage B is further divided into sublineages B1 (B1a, B1b, and B1c) and B2Citation23. In another classification system based on VP4 sequences, CA16 strains isolated between 1999 and 2004 were classified into three lineages, A, B, and CCitation24.
Mutation and recombination are well-recognized mechanisms for the genetic variation and evolution of enterovirusesCitation25,Citation26. The lack of fidelity of the RNA-dependent RNA polymerase of these viruses results in high error rates through nucleotide mis-incorporation during genome replication, explaining how these viruses rapidly mutate and evolveCitation3,Citation27. However, CA16 is relatively genetically stable compared with EV71, with fewer CA16 than EV71 subgenotypes having been identifiedCitation21,Citation26. Viruses from different serotypes, genotypes, and subgenotypes of EV71 and CA16 have been reported to cocirculate and coinfect the same patients during epidemics of HFMD in the Asia-Pacific regionCitation16,Citation28, with intratypic and intertypic genetic recombination among enteroviruses having been frequently reportedCitation11,Citation16,Citation21,Citation23,Citation29–Citation43. Complete genome analyses revealed preferential recombination sites located in the nonstructural protein coding regions P2 (2A/2B junction) and P3 (3C and 3D regions), where nucleotide sequences with high similarities are present among EV-ACitation11,Citation16,Citation21,Citation23,Citation26,Citation31–Citation33. The emergence of recombinant viruses, either recombined between strains within the EV71 serotype or between the EV71 and CA16 serotypes, have been postulated to potentially be responsible for large-scale HFMD outbreaks, and these strains are associated with higher virulence in mouse models and in in vitro assaysCitation11,Citation44,Citation45. The first evidence for intratypic and intertypic recombination in EV71 strains was reported during an outbreak in the late 1990s, which was subsequently followed by confirmatory reports from several countries in East and Southeast AsiaCitation11,Citation16,Citation21,Citation23,Citation26,Citation29–Citation43. In addition, the emergence of EV71 double- or triple-recombinant strains was reported during HFMD epidemics in 2008–2010Citation36,Citation37,Citation39. In this study, the complete genome sequences of EV71 and CA16 isolates obtained between 2006 and 2014 in Thailand were analyzed for subgenotype and genetic recombination. Our results suggest that multiple introductions of recombinant viruses into Thailand are likely to have occurred.
Results
Enteroviral subgenotyping
The VP1 nucleotide sequences from 32 enteroviruses were phylogenetically analyzed together with reference sequences using MEGA version 5.0 (https://www.megasoftware.net). The viruses were identified as 25 EV71 isolates (comprising 20 B5, 1 C2, 2 C4a, and 2 C4b subgenotypes) and 7 CA16 isolates (comprising 6 B1a and 1 B1b subgenotypes). Notably, the dominant EV71 strains circulating between 2011 and 2014 belonged to subgenotype B5. The EV71 subgenotype C4b was first isolated in Thailand in 2006 in this study but was not detected in 2009, whereas the subgenotype C4a was first isolated in 2009 and currently persists in Thailand. CA16 isolates were less commonly detected than EV71 isolates. Of the seven CA16 isolates, six belonged to the B1a subgenotype and one belonged to the B1b subgenotype. Accession numbers for the VP1 nucleotide sequences of these viruses are shown in Table .
List of the 32 enteroviruses, including 25 EV71 and 7 CA16 strains, used for phylogenetic tree construction and genetic recombination analyses
Genomic characterization of the enteroviral isolates
Among the 25 EV71 isolates, only 5 full-length genome sequences were obtained. Excluding the poly(A) tracts, the complete genomes of the four EV71-B5 isolates were 7412 nucleotides long, while that of an EV71-C2 subgenotype isolate was 7408 nucleotides long. The noncoding 5ʹ-UTRs were 747 nucleotides long, while the 3ʹ-UTRs preceding the poly(A) tail were 83 nucleotides long. The 5ʹ-UTRs and/or the 3ʹ-UTRs of the remaining 20 EV71 isolates were not completely sequenced, and genome sequence lengths of 7318–7409 nucleotides were determined. Based on analyses of the full-length genome sequences, the G + C content of the EV71 genomes varied from 47.95–48.31%, excluding the 3ʹ-poly(A) tail.
The complete genome sequence of a CA16 subgenotype B1a isolate was 7410 nucleotides long and was flanked by a 745-nucleotide 5ʹ-UTR and an 82-nucleotide 3ʹ-UTR preceding the poly(A) tail. The genome sequences of the remaining six isolates had lengths between 7371 and 7402 nucleotides. The G + C content of the complete CA16 genome was 47.4%, excluding the 3ʹ-poly(A) tail.
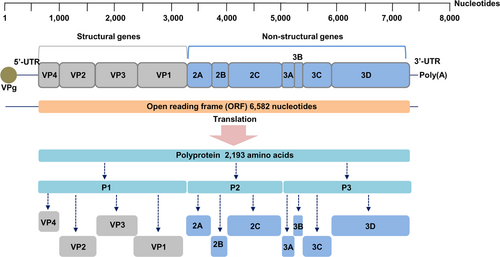
All the EV71 and CA16 genomes contained a single open reading frame (ORF) of 6582 nucleotides encoding a polyprotein of 2193 amino acids as shown in Fig. . All genomic sequences have been deposited in the GenBank database with the accession numbers KX372308 to KX372339.
Fig. 1 Enterovirus RNA genome organization and processing pattern of the polyprotein
Phylogenetic analyses of the enteroviral genomes
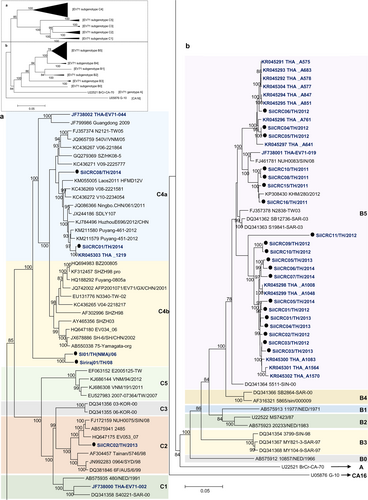
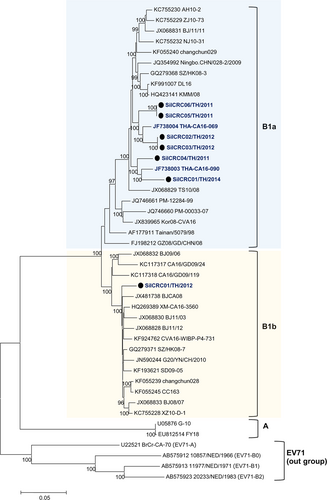
Phylogenetic trees were constructed based on the nucleotide sequences comprising the 5ʹ-UTR and the P1 (VP1–VP4), P2 (2A–2C), and P3 (3A–3D) regions. The nucleotide sequences of the Thai EV71 and CA16 isolates were analyzed for their genetic relationships with the reference subgenotypes, EV-A prototypes and the oldest available strainsCitation41 available in the GenBank database using MEGA 5.0. All of the enterovirus isolates assayed in this study clustered together with their prototypes and the oldest available strains EV-A strains, as shown in Fig. and Supplementary Figure S1. The results identified the 25 EV71 isolates as 20 B5, 2 C4a, 2 C4b, and 1 C2 subgenotypes, while among the seven CA16 isolates, six isolates belonged to the B1a subgenotype while one was a B1b subgenotype as shown in Fig. . A subgenotyping analysis based on the VP1 region or the entire genome yielded the same result.
The phylogenetic tree was extended for a EV71 genotype C and b EV71 genotype B, based on complete genome sequences. The phylogenetic dendrogram was constructed by the neighbor-joining method on the basis of the maximum composition likelihood model using MEGA 5.0. The prototype CA16 G-10 strain was used as an outgroup. Bootstrap values of 1000 replicates are shown at the branch points. EV71 Thai isolates are indicated in blue letters and strains marked by black circles were from this study
The phylogenetic dendrogram was constructed by the neighbor-joining method on the basis of the maximum composition likelihood model using MEGA 5.0. The prototype EV71 genotypes A and B were used outgroups. Bootstrap values of 1000 replicates are shown at the branch points. CA16 Thai isolates are indicated in blue letters and strains marked by black circles were from this study
Analyses for genetic recombination in the viral genomes
The viral genome regions of the study viruses, including the 5ʹ-UTR and the P1, P2, and P3 regions, were phylogenetically analyzed for genetic relationships with the other enteroviral genomes. Similarity plots and bootscan analyses were performed to identify potential genetic recombination sites in the viral genomes using various prototypes and the oldest available EV-A strains as the reference sequences (Supplementary Table S3).
Phylogenetic tree analysis of each viral genome region showed that 19 of 20 EV71-B5 Thai isolates clustered together using the 5511-SIN-00 EV71-B5 prototype as the reference strain (accession number DQ341364). The highest degree of similarity to the reference nucleotide sequence suggested that these 19 EV71-B5 Thai isolates were nonrecombinant strains. In contrast, the P3 region of an EV71-B5 strain, SiICRC11/TH/2012, was more evolutionary distant and was segregated from the reference strain. Similarity plot and bootscan analyses showed the recombination breakpoint at the P2–P3 region at approximate nucleotide positions of 4500 and 5800, suggesting intratypic recombination with EV71 genotype A or intertypic recombination with the other EV-A members as shown in Fig. and Supplementary Figures S2 and S3. In addition, phylogenetic analysis showed that the EV71-C2 isolate SiICRC02/TH/2013 was not a recombinant virus and was closely related to the oldest identified strain Tainan/5746/98 (GenBank accession no. AF304457). In contrast, the two EV71-C4a isolates (SiICRC01/TH/2014 and SiICRC08/TH/2014) and the two EV71-C4b (SI01/TH(NMA)/06 and Siriraj01/TH/08) isolates were closely related to SHZH98, the oldest identified C4 isolate and a previously known recombinant virusCitation29, as shown in Supplementary Figures S1 and S4. A phylogenetic tree of the P1 regions showed that all four EV71-C4 Thai isolates resembled the EV71 genotype C, while the P2 regions resembled EV71 genotype B and the P3 regions resembled the other EV-A members (CA4, CA14, and CA16) (Supplementary Figures S2 and S3). The phylogenetic analysis results agreed well with both the similarity plot and bootscan analyses, which demonstrated the occurrence of double recombination with two breakpoints in the P2 (resembling the EV71 genotype B: MS/7423/87) and the P3 (resembling CA4, CA14, and CA16) regions. This type of double recombination was observed in all EV71-C4 isolates as shown in Fig. and Supplementary Figures S2–S5. The results are summarized in Table .
The similarity was calculated in a sliding window size of 200 nucleotides using the Kimura 2-parameter distance method. Bootscan analysis was performed using the neighbor-joining tree model and the Kimura 2-parameter distance algorithm with a window size of 200 nucleotides
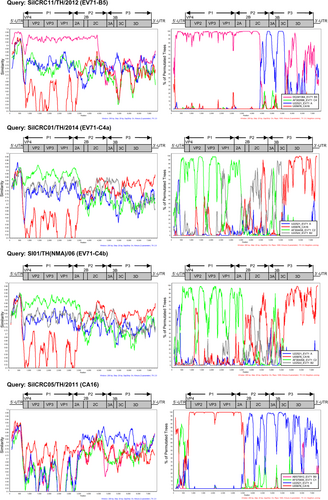
Summary of genetic recombination events among the enterovirus isolates circulating in Thailand between 2006 and 2014
Similarly, the phylogenetic tree, similarity plot and bootscan analyses showed that all seven CA16 strains studied were also recombinant viruses. The P1 region of the viral genomes showed the highest similarity to the CA16 prototype, but the P2 and P3 regions resembled several enteroviruses, including EV71 genotypes A, CA3, CA6, and CA12, suggesting intertypic recombination events. The recombination breakpoints were identified at the 2A–2B junctions (Fig. , Supplementary Figures S2–S6 and Table ).
Analysis of viral genome identity
The genomic sequences of the study viruses were comprehensively analyzed by region, i.e., the 5ʹ-UTR and the P1, VP4, VP2, VP3, VP1, P2, 2A, 2B, 2C, P3, 3A, 3B, 3C, and 3D regions, for identity to the oldest representative strains or prototypes, including EV71-A, EV71-B5, EV71-C2, CA3, CA4, CA6, CA12, CA14, and CA16, as shown in Supplementary Table S4. The nucleotide sequences of 19 nonrecombinant EV71-B5 isolates perfectly matched the reference EV71-B5 strain 5511-SIN-00 for all the analyzed regions, while the recombinant EV71-B5 isolate (SiICRC11/TH/2012) shared high nucleotide sequence identity to the reference EV71-B5, except for the sequences at the recombination breakpoints (the P3, 3A, 3B, and 3C regions) which were closer to EV71-A, CA3, CA6, and CA12. The nucleotide sequences of the EV71-C4a and C4b isolates showed high sequence identities to the reference EV71 genotype C strain Tainan/5746/98, except for the sequences at the recombination breakpoints (the P2, 2B, 2C, 3A, 3B, 3C, and 3D regions) which were closer to EV71-B5, CA4, CA14, and CA16. Similarly, all of the seven CA16 isolates assayed in this study shared a high degree of homology to the G-10 prototype strain, except for the sequences at the recombination breakpoints (the P2, 2B, 2C, P3, 3A, 3B, 3C, and 3D regions) which showed higher similarity to EV71 A, CA3, CA6, and CA12.
Discussion
Multiple subgenotypes of EV71 have circulated in the Thai population during the past two decades, starting with B4 and C1 in 2001 and followed by C5 in 2006, C4b in 2006, B5 and C2 in 2007, and C4a in 2009 (information obtained from the GenBank database sequences deposited by our laboratory and others)Citation21,Citation46–Citation49. Subgenotype replacement was observed over time, but the exact times of their emergence and extinction are not known in most cases. Nevertheless, this study identified SI01/TH(NMA)/06 as the first C4b strain introduced into Thailand in 2006, a virus that was isolated from a child who died of encephalitis. Molecular epidemiology and field investigations suggested that this strain originated from Shenzhen, ChinaCitation46. Our surveillance suggested that the subgenotype C4b disappeared after 2008. An analysis of the EV71 strain THA-EV71-044 (GenBank accession no. JF738002) documented in 2009 revealed it to be the first C4a subgenotype ever reported in ThailandCitation48. While the circulating EV71 strains belong to multiple EV71 subgenotypes, the CA16 strains circulating in Thailand were primarily confined to subgenotype B1a. Only one CA16 B1b isolate, SiICRC01/TH/2012, was detected in 2012. Several other HFMD-associated enteroviruses have occasionally been isolated by our laboratory, such as coxsackieviruses A4 (CA4), CA6, CB1, CB4, and CB5 and echovirus 19. From 2012 to 2015, CA6 caused a large outbreak of mild HFMD in several countries, including Thailand, and became the dominant subgenotype for a few yearsCitation49,Citation50. At present, the most common HFMD-associated viruses are EV71 followed by CA16 and CA6 (unpublished data).
In the present study, a complete genome analysis of 25 EV71 and 7 CA16 strains isolated between 2006 and 2014 was performed to assess possible genetic recombination within the viral genomes. In general, the VP1 sequence has been used to subgenotype enterovirusesCitation51. Subgenotypic identification based on VP1 or entire genome sequences, as was performed in this study and by others, yielded the same resultsCitation52. The full-length genome and the genomic organization of both EV71 and CA16 viruses in this study were similar to those of EV71 and CA16 strains that were previously published or deposited in the GenBank databaseCitation23,Citation42,Citation53, although slight differences in the lengths of the 5ʹ-UTRs and 3ʹ-UTRs were observed. Our study did not observe any insertions or deletions in the protein encoding regions (P1, P2, and P3), suggesting that the viruses contained conserved functional proteins with no frameshift mutations. However, nucleotide substitutions were scattered across the genomes, suggesting that genetic variations among EV71 and CA16 strains are common. Based on the complete genomic sequences, our phylogenetic trees demonstrated that all 32 enteroviral isolates were perfectly clustered with the prototypes and reference strains. Our viral isolates exhibited an evolutionary relationship with those previously reported from China, Japan, and many countries in Southeast AsiaCitation15,Citation32,Citation43,Citation54. The cocirculation of EV71 and CA16 in the same HFMD outbreak is common and has been observed in various countriesCitation13,Citation16,Citation28, including ThailandCitation47,Citation48.
The oldest identified EV71-B5 strain (5511-SIN-00) was identified in 2000 as a nonrecombinant virus in Singapore. Evidence of genetic recombination in EV71-B5 was first reported in Chongqing City, China, in 2014 in the viral strain CQ2014-86/CQ/CHN/2014 (accession number KU647000), and the recombination breakpoints were in the 5ʹ-UTR and in the P2-P3 regionCitation32. Interestingly, genetic recombination in the EV71-B5 isolate assayed in this study was discovered in 2012. Similarity plot and bootscan analyses revealed that SiICRC11/TH/2012 was the only isolate among the 20 assayed EV71-B5 isolates with the potential for intratypic or intertypic recombination in the 2C region between the nucleotide positions 4500 and 5800, resembling the EV71 genotypes A, CA3, CA6, and CA12. Moreover, the results of our study suggested that all of our two EV71-C4a and two EV71-C4b isolates were double recombinants, with two recombination breakpoints in the 2A–2B junction and the 3C–3D region. Our analysis also showed that this recombination pattern was also present in THA_A1219, an EV71-C4 strain isolated in 2014 by another group of Thai investigatorsCitation43.
We showed that six out of seven CA16 strains in this study belong to subgenotype B1a, with only one strain belonging to subgenotype B1b. The CA16 recombinant viruses may have circulated in Thailand for specific period of time as has occurred in ChinaCitation16,Citation23,Citation30. The results of similarity plot and bootscan analyses demonstrated multiple intertypic recombinations in all of these seven CA16 strains such that the P1 region resembled the CA16-G10 prototype, but the P2–P3 regions resembled the EV71 genotypes A, CA3, CA6, and CA12. Intertypic recombination in CA16 viruses has been previously reported in mainland ChinaCitation16,Citation23,Citation30. The recombination breakpoint was frequently detected in the P2–P3 regions, but not in the P1 region, which encodes the VP1–VP4 structural proteins. This finding suggests that the nonstructural proteins of enterovirus members are highly conserved and are interchangeable.
At present, HFMD is not only confined to the Asia-Pacific region. HFMD epidemics associated with subgenotypes other than B5 and C4 (primarily from C1 and C2) have been reported from several countries in Europe and North AmericaCitation3,Citation4,Citation21,Citation55–Citation58. Genetic recombination was also observed in those viruses. However, HFMD associated with recombinant EV71 or C16 viruses is more common in Asian countries, although the role of these recombinant viruses in public health is uncertain. Clinical manifestations in patients infected with recombinant and nonrecombinant EV71 isolates were comparable, suggesting that the virulence of these types of strains might not be differentCitation41. In contrast, the results of viral phenotype studies in cell culture- and mouse model-based systems suggested that the recombinant viruses are likely more virulentCitation44,Citation45. The role of recombinant viruses with respect to the magnitude of an HFMD outbreak is also controversial. A large outbreak involving 6049 HFMD cases in Fuyang, China, was caused by EV71-C4a together with a small number of intertypic recombinant EV71-C4a viruses exhibiting CA16 G-10 sequences in the 3D regionCitation11. Similarly, a small number of intratypic EV71-C4a recombinant viruses with EV71-C4b sequences at the 2B–2C region were isolated in mainland China during a very large outbreak between 2011 and 2012 that involved millions of HFMD casesCitation16. The HFMD outbreak in Cambodia in 2012, which resulted in 61 cases with 56 deaths, was primarily caused by EV71-C4a and few EV71-B5 subgenotypesCitation15.
The impact of the emergence of recombinant viruses in HFMD will require a great deal of observation. The recombinant EV71-C4a strain was predominantly found in China in 2008Citation11, and is currently the major circulating virus. Subsequently, this recombinant virus may have spread to several other countries. EV71-B5 has become the predominant subgenotype associated with HFMD in Thailand in recent years. The role of the EV71-B5 recombinant virus on the spread of HFMD and clinical outcomes should be further explored. Moreover, surveillance for circulating strains and subgenotype replacement are also important in terms of vaccine selection and development.
Materials and methods
Ethics issue
This study was approved by the Siriraj Hospital Institutional Review Board, Faculty of Medicine, Siriraj Hospital, Mahidol University, with the certificate of approval number Si058/2013 for the protocol number 689/2555(EC3).
Cell line
Vero cells (ATCC, catalog no. CCL-81) derived from African green monkey kidney were cultured in Earle’s minimal essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, NY) penicillin, gentamycin, and fungizone. The cell monolayers used for viral isolation and propagation were maintained in media supplemented with 2% FBS.
Study viruses
Throat swabs and fecal specimens were collected between 2006 and 2014 from various provinces of Thailand, and a total of 32 enteroviruses were isolated from 31 pediatric patients (30 HFMD and 1 encephalitis case) and 1 asymptomatic adult with a history of contact with an HFMD patient. Information for some patients has been reported previouslyCitation46,Citation47. The viruses were grown for 2 or 3 passages before being subjected to nucleotide sequencing.
Polymerase chain reaction (PCR) and complete genome sequencing
Culture supernatants of enterovirus-infected Vero cells were extracted for total RNA using a QIAamp® Viral RNA mini kit (Qiagen, GmbH, Hilden, Germany). The viral RNA was amplified in eight DNA fragments that spanned the entire genome (~7.4 kilobases (kb) long). Each DNA fragment was sequenced directly using a panel of sequencing primers designed by our group and previous investigatorsCitation53 as listed in Supplementary Tables S1 and S2.
One step RT-PCR kits (Qiagen GmbH, Hilden, Germany) were used for viral RNA amplification following the manufacturer’s instructions. A 50 μl reaction mixture was subjected to reverse transcription at 50 °C for 30 min, followed by heat inactivation of the reverse transcriptase enzyme together with activation of DNA polymerase at 95 °C for 15 min that was followed by 35 cycles of denaturation at 94 °C for 45 s, primer annealing at 48–55°C for 45 s and extension at 72 °C for 2 min. The reaction was completed with a final extension step at 72 °C for 10 min. The PCR DNA products were electrophoresed, purified using a QIAquick® gel extraction kit (Qiagen), and were sequenced by Macrogen Inc., Seoul, Korea (http://dna.macrogen.com/eng/). Bidirectional sequences were obtained and assembled using BioEdit Sequence Alignment Editor version 7.0.4.1Citation59. Overlapping DNA sequences with at least 85% homology and a minimum of 20 nucleotides of overlap were CAP contiged to generate consensus sequences.
Nucleotide sequencing and phylogenetic tree construction
Nucleotide sequences of the viral genomes and the deduced amino acid sequences were analyzed using BioEdit and the Basic Local Alignment Search Tool (BLAST) available at the U.S. National Center of Biotechnology Information (NCBI). The complete nucleotide sequences of all 32 enterovirus strains have been deposited in the GenBank database (www.ncbi.nlm.nih.gov) under accession numbers KX372308 to KX372332 for EV71 and KX372333 to KX372339 for CA16, as shown in Table .
The nucleotide sequences encompassing the entire genomes or particular regions (5ʹ-UTR, P1, VP4, VP2, VP3, VP1, P2, 2A, 2B, 2C, P3, 3A, 3B, 3C, or 3D) were aligned with reference sequences retrieved from the GenBank database using the ClustalW multiple alignment application. Phylogenetic trees were constructed using MEGA version 5.0 (https://www.megasoftware.net/)Citation60. The evolutionary distances were estimated using the neighbor-joining method and a maximum composite likelihood algorithm. The reliability of the neighbor-joining tree was estimated by a bootstrapping analysis using 1000 replicate datasets. The reference sequences of CA16 were used as outgroups for EV71 phylogenetic tree construction, while the EV71 genotypes A and B were used as the outgroups for CA16 phylogenetic tree construction. Bootstrap values of greater than 80% supported the tree topology, as shown at the nodes of each cluster.
Genetic recombination analysis
The 32 enteroviral genomes assayed in this study were investigated for intratypic and intertypic genetic recombination. The genomic sequences were aligned with the prototypes and the oldest available strains of the species Enterovirus A (EV-A) using the ClustalW multiple alignment application in BioEdit. Sequences of the reference viral genomes were retrieved from the GenBank database using the accession numbers listed in Supplementary Table S3.
Similarity plot and bootscan analyses were performed using SimPlot (Similarity Plotting) version 3.5.1 (Stuart Ray, Johns Hopkins University, Baltimore, MD) (http://sray.med.som.jhmi.edu/SCRoftware/simplot/)Citation61 using the default parameter settings. All gaps were removed from the DNA sequences, and ≥ 50% nongaps were required to analyze a set of data points. Bootscan analysis was performed using the neighbor-joining tree model and the Kimura 2-parameter distance algorithm with a window size of 200 nucleotides moving along the alignment in increments of 20 nucleotides with 1000 resampling. The use of the PHYLIP internal code (v3.5) and a 70% parental threshold were selected for notification if a region of recombination was detected. The nucleotide positions were indicated along the x-axis, and the sequence similarities or percentages of permutated trees were indicated along the y-axis.
Supplementary Figure S6
Download TIFF Image (4.7 MB)Supplementary Table S1
Download MS Word (49 KB)Supplementary Table S2
Download MS Word (49 KB)Supplementary Table S3
Download MS Word (51 KB)Supplementary Table S4
Download MS Word (179 KB)Supplementary Figure S1
Download TIFF Image (1.5 MB)Supplementary Figure S2
Download TIFF Image (2.9 MB)Supplementary Figure S3
Download TIFF Image (3 MB)Supplementary Figure S4
Download TIFF Image (2.6 MB)Supplementary Figure S5
Download TIFF Image (5.8 MB)Acknowledgements
This work was supported by the National Science and Technology Development Agency. The authors would like to thank Prof. Duncan R. Smith from the Institute of Molecular Biosciences, Mahidol University for English editing of this paper.
Author contributions
P.P. conceived and designed the research study and wrote the paper; P.N. performed the experiments, analyzed the data, and wrote the paper; K.S. performed the experiments and analyzed the data; J.P. and C.K. performed the experiments; S.P. analyzed the data; R.B., A.M., A.T., and K.C. provided the clinical specimens for viral isolation. All authors read and approved the final version of the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41426-018-0215-x).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related Research Data
References
- EspositoSPrincipiNHand, foot and mouth disease: current knowledge on clinical manifestations, epidemiology, aetiology and preventionEur. J. Clin. Microbiol Infect. Dis.201837 391 39810.1007/s10096-018-3206-x
- KashyapRRKashyapRSHand, foot and mouth disease - a short case reportJ. Clin. Exp. Dent.20157e336e33810.4317/jced.52031
- LeiXCuiSZhaoZet al.Etiology, pathogenesis, antivirals and vaccines of hand, foot, and mouth diseaseNatl. Sci. Rev.2015226828410.1093/nsr/nwv038
- SolomonTLewthwaitePPereraDet al.Virology, epidemiology, pathogenesis, and control of enterovirus 71Lancet Infect. Dis.20101077879010.1016/S1473-3099(10)70194-8
- ChanLGParasharUDLyeMSet al.Deaths of children during an outbreak of hand, foot, and mouth disease in Sarawak, Malaysia: clinical and pathological characteristics of the disease. For the outbreak Study GroupClin. Infect. Dis.20003167868310.1086/314032
- AbuBakarSCheeHYAl-KobaisiMFet al.Identification of enterovirus 71 isolates from an outbreak of hand, foot and mouth disease (HFMD) with fatal cases of encephalomyelitis in MalaysiaVirus Res1999611910.1016/S0168-1702(99)00019-2
- LinTYTwuSJHoMSet al.Enterovirus 71 outbreaks, Taiwan: occurrence and recognitionEmerg. Infect. Dis.2003929129310.3201/eid0903.020285
- LinTYChangLYHsiaSHet al.The1998 Enterovirus 71 outbreak in Taiwan: pathogenesis and managementClin. Infect. Dis.200234Suppl 2S52S5710.1086/338819
- ZhuangZCKouZQBaiYJet al.Epidemiological research on hand, foot, and mouth disease in mainland ChinaViruses201576400641110.3390/v7122947
- WangYFengZYangYet al.Hand, foot, and mouth disease in China: patterns of spread and transmissibility during 2008-2009Epidemiology20112278179210.1097/EDE.0b013e318231d67a
- ZhangYZhuZYangWet al.An emerging recombinant human enterovirus 71 responsible for the 2008 outbreak of hand foot and mouth disease in Fuyang city of ChinaVirol. J.201079410.1186/1743-422X-7-94
- NguyenNTPhamHVHoangCQet al.Epidemiological and clinical characteristics of children who died from hand, foot and mouth disease in Vietnam, 2011BMC Infect. Dis.20141410.1186/1471-2334-14-341
- VanTuPThaoNTTPereraDet al.Epidemiologic and virologic investigation of hand, foot, and mouth disease, southern Vietnam, 2005Emerg. Infect. Dis.2007131733174110.3201/eid1311.070632
- SeiffACambodia unravels cause of mystery illnessLancet201238020510.1016/S0140-6736(12)61200-8
- DuongVMeyCEloitMet al.Molecular epidemiology of human enterovirus 71 at the origin of an epidemic of fatal hand, foot and mouth disease cases in CambodiaEmerg. Microbes Infect.2016510.1038/emi.2016.101
- LiuWWuSXiongYet al.Co-circulation and genomic recombination of coxsackievirus A16 and enterovirus 71 during a large outbreak of hand, foot, and mouth disease in Central ChinaPLoS One20149e9605110.1371/journal.pone.0096051
- ChangLYLinTYHuangYCet al.Comparison of enterovirus 71 and coxsackievirus A16 clinical illnesses during the Taiwan enterovirus epidemic, 1998Pediatr. Infect. Dis. J.1999181092109610.1097/00006454-199912000-00013
- LeeKYEnterovirus 71 infection and neurological complicationsKorean J. Pediatr.20165939540110.3345/kjp.2016.59.10.395
- ChenLHeYQMengJet al.Full-genome sequences of seven fatal enterovirus 71 strains isolated in Shenzhen, China, in 2014Genome Announc20164e00316e003164850858
- LinKZhangHBaiWet al.Complete genome sequence of an enterovirus 71 strain isolated from the cerebrospinal fluid of a child with severe hand-foot-and-mouth disease in Yunnan, China, 2013Genome Announc20175e00274175427203
- YipCCLauSKWooPCet al.Human enterovirus 71 epidemics: what’s next?Emerg. Health Threats J.201361978010.3402/ehtj.v6i0.19780
- BrownBAObersteMSAlexanderJPet al.Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998J. Virol.19997399699975113047
- ChenXTanXLiJet al.Molecular epidemiology of coxsackievirus A16: intratype and prevalent intertype recombination identifiedPLoS One20138e8286110.1371/journal.pone.0082861
- LiLHeYYangHet al.Genetic characteristics of human enterovirus 71 and coxsackievirus A16 circulating from 1999 to 2004 in Shenzhen, People’s Republic of ChinaJ. Clin. Microbiol2005433835383910.1128/JCM.43.8.3835-3839.2005
- LukashevANLashkevichVAIvanovaOEet al.Recombination in circulating enterovirusesJ. Virol.200377104231043110.1128/JVI.77.19.10423-10431.2003
- HuangSWKiangDSmithDJet al.Evolution of re-emergent virus and its impact on enterovirus 71 epidemicsExp. Biol. Med (Maywood)201123689990810.1258/ebm.2010.010233
- WoodmanAArnoldJJCameronCEet al.Biochemical and genetic analysis of the role of the viral polymerase in enterovirus recombinationNucleic Acids Res2016446883689510.1093/nar/gkw567
- ZhangCZhuRYangYet al.Phylogenetic analysis of the major causative agents of hand, foot and mouth disease in Suzhou city, Jiangsu province, China, in 2012-2013Emerg. Microbes Infect.2015410.1038/emi.2015.12
- Yoke-FunCAbuBakarSPhylogenetic evidence for inter-typic recombination in the emergence of human enterovirus 71 subgenotypesBMC Microbiol200667410.1186/1471-2180-6-74
- ZhaoKHanXWangGet al.Circulating coxsackievirus A16 identified as recombinant type A human enterovirus, ChinaEmerg. Infect. Dis.201117153715403381541
- ChanYFAbuBakerSRecombinant human enterovirus 71 in hand, foot and mouth disease patientsEmerg. Infect. Dis.2004101468147010.3201/eid1008.040059
- YangQZhangYYanDet al.Isolation of an imported subgenotypes B5 strain of human enterovirus A71 in Chongqing City, China, 2014Virol. J.20161311510.1186/s12985-016-0571-x
- LiJHuoXDaiYet al.Evidences for intertypic and intratypic recombinant events in EV71 of hand, foot and mouth disease during an epidemic in Hubei Province, China, 2011Virus Res201216919520210.1016/j.virusres.2012.07.028
- Thoa lePKChiangPSKhanhTHet al.Genetic and antigenic characterization of enterovirus 71 in Ho Chi Minh City, Vietnam, 2011PLoS One20138e6989510.1371/journal.pone.0069895
- HuangSWHsuYWSmithDJet al.Reemergence of enterovirus 71 in 2008 in Taiwan: dynamics of genetic and antigenic evolution from 1998 to 2008J. Clin. Microbiol2009473653366210.1128/JCM.00630-09
- YipCCLauSKZhouBet al.Emergence of enterovirus 71 “double-recombinant” strains belonging to a novel genotype D originating from southern China: first evidence for combination of intratypic and intertypic recombination events in EV71Arch. Virol.20101551413142410.1007/s00705-010-0722-0
- YipCCLauSKLoJYet al.Genetic characterization of EV71 isolates from 2004 to 2010 reveals predominance and persistent circulation of the newly proposed genotype D and recent emergence of a distinct lineage of subgenotype C2 in Hong KongVirol. J.20131022210.1186/1743-422X-10-222
- GuoWPLinXDChenYPet al.Fourteen types of co-circulating recombinant enterovirus were associated with hand, foot, and mouth disease in children from Wenzhou, ChinaJ. Clin. Virol.201570293810.1016/j.jcv.2015.06.093
- LiuYZhangFFuCet al.Combination of intratypic and intertypic recombinant events in EV71: a novel evidence for the “triple-recombinant” strains of genotype A viruses in Mainland China from 2008 to 2010Virus Genes20155036537410.1007/s11262-015-1170-4
- DingNZWangXMSunSWet al.Appearance of mosaic enterovirus 71 in the 2008 outbreak of ChinaVirus Res200914515716110.1016/j.virusres.2009.06.006
- HuangSCHsuYWWangHCet al.Appearance of intratypic recombination of enterovirus 71 in Taiwan from 2002 to 2005Virus Res200813125025910.1016/j.virusres.2007.10.002
- ZhangYTanXCuiAet al.Complete genome analysis of the C4 subgenotype strains of enterovirus 71: predominant recombination C4 viruses persistently circulating in China for 14 yearsPLoS One20138e5634110.1371/journal.pone.0056341
- MauleekoonphairojJVongpunsawadSPuenpaJet al.Complete genome sequence analysis of enterovirus 71 isolated from children with hand, foot, and mouth disease in Thailand, 2012-2014Virus Genes20155129029310.1007/s11262-015-1239-0
- PhuektesPChuaBHSandersSet al.Mapping genetic determinants of the cell-culture growth phenotype of enterovirus 71J. Gen. Virol.2011921380139010.1099/vir.0.029371-0
- WeiWGuoHLiJet al.Circulating HFMD-associated coxsackievirus A16 is genetically and phenotypically distinct from the prototype CV-A16PLoS One20149e9474610.1371/journal.pone.0094746
- BuathongRHanshoaworakulWSutdanDet al.Cluster of fatal cardiopulmonary failure among children caused by an emerging strain of enterovirus 71, Nakhorn Ratchasima Province, ThailandOutbreak Surveill. Investig. Rep.2008113
- PiriyapornpipatSPittayawonganonCMungaomklangAet al.Investigation of a severe enterovirus encephalitis and circulating genotypes during hand, foot and mouth disease surge in Nakhon Ratchasima Province, Thailand, August 2011Outbreak Surveill. Investig. Rep.201471622
- PuenpaJTheamboonlersAKorkongSet al.Molecular characterization and complete genome analysis of human enterovirus 71 and coxsackievirus A16 from children with hand, foot and mouth disease in Thailand during 2008-2011Arch. Virol.20111562007201310.1007/s00705-011-1098-5
- PuenpaJChieochansinTLinsuwanonPet al.Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012Emerg. Infect. Dis.20131964164310.3201/eid1904.121666
- ChatproedpraiSTemparkTWanlapakornNet al.Unusual skin manifestation of hand, foot and mouth disease associated with coxsackievirus A6: case report+201544504872
- CardosaMJPereraDBrownBAet al.Molecular epidemiology of human enterovirus 71 strains and recent outbreaks in the Asia-Pacific region: comparative analysis of the VP1 and VP4 genesEmerg. Infect. Dis.2003946246810.3201/eid0904.020395
- ChanYFSamICAbuBakarSPhylogenetic designation of enterovirus 71 genotypes and subgenotypes using complete genome sequencesInfect. Genet Evol.20121040441210.1016/j.meegid.2009.05.010
- ChangSCLiWCChenGWet al.Genetic characterization of enterovirus 71 isolated from patients with severe disease by comparative analysis of complete genomesJ. Med Virol.20128493193910.1002/jmv.23287
- DonatoCHoi leTHoaNTet al.Genetic characterization of enterovirus 71 strains circulating in Vietnam in 2012Virology20164951910.1016/j.virol.2016.04.026
- HasselCMirandALukashevAet al.Transmission patterns of human enterovirus 71 to, from and among European countries, 2003 to 2013Eur. Surveill.2015203000510.2807/1560-7917.ES.2015.20.34.30005
- MirandASchuffeneckerIHenquellCet al.Phylogenetic evidence for a recent spread of two populations of human enterovirus 71 in European countriesJ. Gen. Virol.2010912263227710.1099/vir.0.021741-0
- van der SandenSvan EekJMartinDPet al.Detection of recombination breakpoints in the genomes of human enterovirus 71 strains isolated in the Netherlands in epidemic and non-epidemic years, 1963-2010Infect. Genet Evol.20111188689410.1016/j.meegid.2011.02.011
- van der SandenSKoopmansMUsluGet al.Epidemiology of enterovirus 71 in the Netherlands, 1963 to 2008J. Clin. Microbiol2009472826283310.1128/JCM.00507-09
- HallTABioEdit: a user-friendly biological sequence alignment editor and analysis program for Window 95/98/NTNucleic Acids Symp . Ser.1999419598
- TamuraKPetersonDPetersonNet al.MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methodsMol. Biol. Evol.2011282731273910.1093/molbev/msr121
- LoleKSBollingerRCParanjapeRSet al.Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombinationJ. Virol.199973152160103818