
Abstract
Background
High-throughput sequencing of genes indicating susceptibility to noise-induced hearing loss has not previously been reported.
Aims/Objectives
To identify and analyze genes associated with susceptibility to noise-induced hearing loss (NIHL) and characterize differences in susceptibility to hearing loss by genotype.
Material and methods
Pure tone audiometry tests were performed on 113 workers exposed to high-intensity noise. Whole-exome sequencing (WES) was conducted and NIHL susceptibility genes screened for training unsupervised and supervised machine learning models. Immunofluorescence staining of mouse cochlea was used to observe patterns of NIHL susceptibility gene expression.
Results
Participants were divided into a NIHL and a control group, according to the results of audiometry tests. Seventy-three possible NIHL susceptibility genes were input into the machine learning model. Two subgroups of NIHL could be distinguished by unsupervised machine learning and the classification was evaluated by the supervised machine learning algorithm. The VWF gene had the highest mutation frequency in the NIHL group and was expressed mainly in the spiral ligament.
Conclusions and significance
NIHL susceptibility genes were screened and NIHL subgroups could be distinguished. VWF may be a novel NIHL susceptibility gene.
Chinese Abstract
背景:标示对噪声诱导听力损失易感性的基因高通量测序以前未被报告过。
目的:识别和分析与噪声诱导听力损失易感性相关的基因(NIHL) 并描述不同基因型的对听力损失易感性的差异。
材料和方法:对 113 名暴露于高强度噪声的工人进行了纯音听力测试。 为了训练无监督和监督机器学习模型, 进行了全外显子组测序(WES)和 NIHL 易感基因筛选。 小鼠耳蜗的免疫荧光染色用于观察 NIHL 易感基因表达模式。
结果:根据听力测试结果, 参与者被分为 NIHL 组和对照组。 将 73 个可能的 NIHL 易感基因输入机器学习模型。 可以通过无监督机器学习区分 NIHL 的两个亚群, 并通过监督机器学习算法来评估分类。NIHL组的 VWF基因突变频率最高, 主要表达于螺旋韧带。
结论及意义:筛选出NIHL易感基因, NIHL亚群可得以区分。 VWF可能是一种新的NIHL易感基因。
Introduction
Hearing loss and tinnitus are common adverse consequences of exposure to high-intensity noise [Citation1]. Long-term exposure causes damage to the auditory system which first results in hearing loss at high frequencies and then gradually extends to speech frequencies [Citation2]. Genetic variation influences noise sensitivity and results in a spectrum of noise-induced hearing loss (NIHL) for people exposed to the same working environment. NIHL is complex and involves environmental and genetic factors, the latter of which may account for about 50% of cases. Over 300 genes distributed on 23 pairs of human chromosomes have been linked to hereditary hearing loss but only 140 genes have been identified. Over 30 gene variants have been associated with NIHL as a result of animal studies or human epidemiological investigations [Citation3]. Previous genetic studies have focused on a few candidate single nucleotide polymorphisms (SNP) at a limited number of sites [Citation4]. Non-high-throughput analysis methods fail to identify new susceptibility genes from the whole genome or the whole exome.
Whole-exome sequencing (WES) is a high-throughput sequencing method at the genome level which sequences all exons throughout the genome using sequence capture technology. WES analysis allows the detection of mutations in gene coding regions to enable the study of pathogenesis at the DNA level. WES has been widely used to identify deafness genes in families but has not previously been applied to genetic variations in the NIHL population.
Machine learning algorithms have been used to analyze WES big data results in the context of disease susceptibility genes [Citation5]. Machine learning methods allow the processing of high-dimensional data sets with many features, such as multiple genes in genome data, as feature input models [Citation6]. A machine learning NIHL gene analysis model enables exploration of disease causes and therapeutic targets and informs noise protection measures and screening of workers exposed to long-term noise.
Unsupervised machine learning algorithms and supervised machine learning methods were used to analyze WES results of workers with long-term noise exposure during the current study. The results are intended to facilitate genetic NIHL screening of susceptible populations with long-term noise exposure.
Materials and methods
Participants and hearing test
Baseline data and hearing status of 113 workers who had experienced long-term exposure of more than one year to an impulse noise engine operating field were retrospectively analyzed. Age, sex and years of noise exposure were collated. Details of medical history of other diseases that may damage hearing were given by participants prior to the hearing test. Pure tone audiometry (Astera, Denmark) at frequencies of 250 Hz, 500 Hz, 1 kHz, 2 kHz, 3 kHz, 4 kHz, 6 kHz and 8 kHz was used to test hearing function.
Subjects with a pure tone audiometry threshold >25dB were considered to have hearing loss and assigned to the NIHL group. Subjects who had a >1 year history of noise exposure but who had a pure tone audiometry threshold ≤25dB at any frequency between 250 and 8 kHz were considered to have no hearing loss and were assigned to the control group. Impulse noise testing was conducted at various locations with different impulse noise engines. A hand-held acoustic analyzer (Denmark B&K Company, BK2250) was used to conduct on-site testing of acoustic parameters at the subjects’ workplaces. Seventeen tests were completed to assess impulse noise at different positions on the test site and the average peak sound pressure level calculated.
Whole-exome sequencing
Peripheral blood was collected from all participants and DNA isolated. Specific enrichment of human whole-exome DNA was performed and high-throughput, high-depth sequencing carried out on HiSeq X1. PE150 sequencing was performed using the Illumina X10 platform (iGenetech, Beijing, China). Exons were double-terminal sequenced, producing a mean read length of 2 × 150 bp and a sequencing depth of 100 ×. High-quality clean reads were obtained from raw data and information analysis carried out with reference to the UCSC genome (hg19, 2009). BWA software was used to compare sequencing data with the reference genome and sequencing was performed simultaneously with repeat sequence removal to obtain the BAM result file. SNPs and indels were identified by SAMtools, GATK and other software and annotated with ANOVAR software to give information on genes, functions and mutation harmfulness [Citation7]. Genomic location, mutation frequency, genotype heterozygosity and pathway of variation were determined for SNPs using external databases. SNP screening criteria included: (1) exon region; (2) nonsynonymous mutation; (3) allele frequency of <0.01 in the East Asian group of ExAC, genomAD and 1000-genome database; (4) harmful prediction for mutation from SIFT, GERP, Polyphen2, Mutation Taster and CADD software. After obtaining information about the mutated genes through WES, Sanger sequencing was performed on the gene with the highest frequency of mutation to verify the existence of the mutation sites.
Machine learning algorithm
Gene mutations detected in at least 5% of NIHL samples were regarded as specific to NIHL. Subjects with NIHL were stratified by genotype to identify subtypes. Distance values were calculated for subjects with NIHL using the Jaccard coefficient [Citation8] and used to group subjects through cluster analysis, the unsupervised machine learning algorithm. A gene-based classification prediction model was constructed by supervised machine learning models, Support Vector Machines (SVM), Random Forest (RF), Multilayer Perceptron (MLP) and eXtreme Gradient Boosting (XGBoost). NIHL patients were randomly divided into training (60%) and test sets (40%) and the former used to train the machine learning algorithms by 10-fold cross-validation. A score for each feature, according to the mean decrease impurity (MDI) which represents the amount of information contained in the population grouping by mutant gene, was assigned by random forest. The importance of variables was ranked using the information gain [Citation9]. Machine learning algorithms were implemented with R (4.2.1).
Machine learning indicators, accuracy, sensitivity, specificity and AUC, were used to evaluate model performance in the test set to compare the prediction performance of different models [Citation10]. ROC curves were also used to evaluate performance of the machine learning model in the test set. The area under the ROC curve (AUC) was used to assess discriminatory power.
Immunofluorescence staining of mouse cochlea
Immunofluorescence staining of 6-week-old c57BL/6 mouse cochlea was performed to assess pathophysiological mechanisms of NIHL susceptibility genes. Mice were sacrificed and cochlear tissue separated, the round window and oval window were opened and fixed in 4% paraformaldehyde at room temperature for 24 h. Samples were decalcified in EDTA for 7 days and frozen sections prepared along the cochlear axis. Sections were sealed in 10% goat serum at 37 °C, incubated with rabbit anti-VWF antibody (Protentech) at 4 °C overnight and with goat anti-rabbit IgG secondary antibody (Protentech) at room temperature for 1 h. Sections were stained with DAPI, an anti-fluorescence quenching sealing agent added and observed under the fluorescence microscope.
Results
Baseline clinical characteristics
Seventy-eight subjects who had experienced long-term noise exposure of >1 year in an engine working environment met the pure tone audiometry criteria for NIHL. Thirty-five subjects who had also been exposed to noise for >1 year without hearing loss were assigned to the control group. All subjects had reported normal hearing prior to noise exposure. No differences in age or working time were present between the NIHL and control groups (). Due to the working environment, all subjects were male. Significant differences in hearing threshold at each frequency were found between the NIHL and control groups (p <.05, , ). On-site noise tests showed that workers’ ears received impulse noise with a mean peak sound pressure level of 141.7 dB from the running of engines in the workplace.
Figure 1. Box diagram visualizes the comparison of pure tone audiometry results between the NIHL group and the control group. The p-values of the independent sample t-tests for two groups at each frequency are labelled.
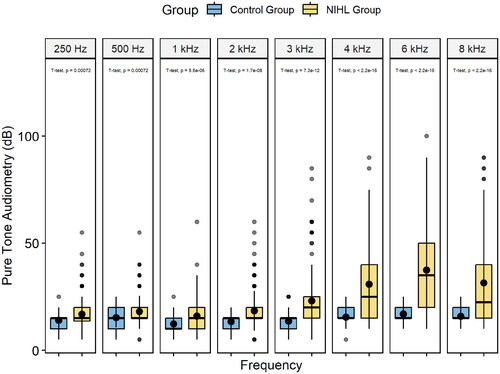
Table 1. Age and work time comparison between the NIHL group and the control group.
Table 2. Comparison of pure tone audiometry results between the NIHL group and the control group.
Analysis of susceptibility genes
Coverage and average depth were calculated after comparison with the original sequence in the human genome (hg19). Coverage rate was >99% for most samples (mean= 99.4%) and sequence depth range was 20 × to 100 ×. Mean target area depth was 44.8.
Mutations with a frequency of less than 0.01 in the genome database and missense mutations predicted to be harmful by protein harmfulness prediction software were selected. Mutations in 73 genes that were detected in at least 5% (4 out of 78) of NIHL samples were used to build machine learning models. No mutations were found in any control sample. Cluster analysis of mutated genes allowed the identification of two NIHL subgroups, A and B, which were distinct from the control group (). The cluster analysis dendrogram shows that subgroup A had 34 members and subgroup B had 44 members. The 78 NIHL subjects could be divided into two different genome maps, according to the presence of mutated genes. Subgroup sensitivity and specificity were analyzed by support vector machines (SVM), random forests (RF), multi-layer perceptron (MLP) and eXtreme Gradient Boosting (XGBoost), using supervised machine learning methods. shows classifier performance and indicates the superiority of MLP for sensitivity, specificity, accuracy and AUC. MLP analysis produced the following values: accuracy 95% CI= 0.742 (0.554, 0.881), sensitivity = 0.692, specificity = 0.778 and AUC = 0.752 (ROC curves shown in ). SVM and RF had equivalent performances to one another for accuracy, sensitivity and specificity while XGBoost showed a poor performance. Hearing status of patients in subgroups A and B were compared and a difference found in high-frequency hearing thresholds (). The difference between the two subgroups was only significant at 3 kHz (p <.05). Thresholds at 4 kHz, 6 kHz and high frequency had a tendency to be different (0.05<p < .1) and the failure to achieve statistical significance may be due to the small sample size or short noise exposure time. In general, the high-frequency hearing threshold of subgroup A was higher than that of subgroup B. Subgroup A was characterized by the presence of mutations in the susceptibility genes, VWF, COL5A1, COL17A1 and MYO18A. Members of subgroup A who had these gene mutations had poorer hearing than members of subgroup B who did not. Therefore, the indications are that the presence of these gene mutations may have contributed to hearing loss due to a noisy environment in members of subgroup A. The principal susceptibility genes for stratification with importance values calculated by MDI in the random forest model shown in brackets were: UTP20 (0.238), DNAH1 (0.225), ADAMTS5 (0.211), SMPD4 (0.211), COL5A1 (0.204), SLC12A6 (0.197), LIPH (0.196), MYO18A (0.193), CRB2 (0.181) and SRMS (0.181). Cluster analysis and classification prediction confirmed that the most significant susceptibility genes distinguishing subgroup A were VWF, SLC12A3, COL17A1 and CPTP and those distinguishing subgroup B were UTP20, ADAMTS5 and ABCB5 (). The susceptibility genes present in subgroup A were associated with poorer hearing and greater harm resulting from noise exposure.
Figure 2. Cluster analysis of 78 noise-induced hearing loss staff. The two subgroups identified by cluster analysis were 34 individuals in subgroup A and 44 individuals in subgroup B.
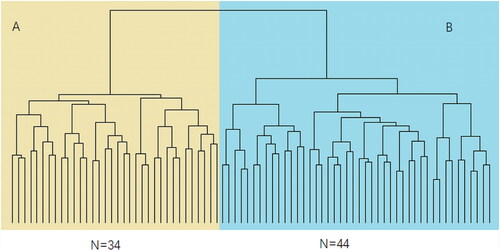
Figure 3. The ROC curves by four machine-learning algorithms.
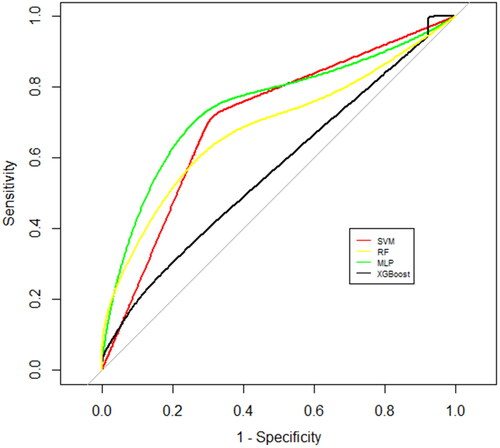
Figure 4. Histogram of gene frequencies after clustering into two subgroups. The two-colored columns represent the mutation numbers of a gene in both subgroups.
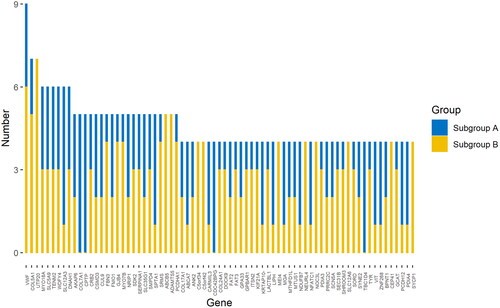
Table 3. Performance of the machine learning algorithms.
Table 4. Pure tone audiometry results of subgroup A and subgroup B.
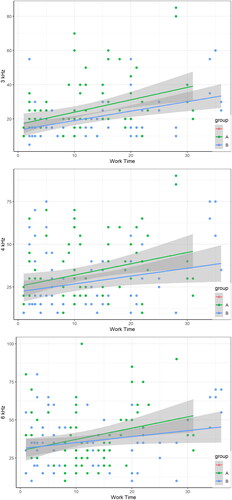
demonstrates that increased numbers of working hours contributed to worsening of hearing. Subgroup A had poorer hearing at frequencies of 3 and 4 kHz than subgroup B. In addition, a shorter working time correlated with worse hearing of subgroup A members when compared with subgroup B. A short working time and a frequency of 6 kHz produced similar results for the hearing of members of both subgroups. Prolongation of the working time correlated with greater deterioration in the hearing of subgroup A members than of subgroup B.
Figure 5. Relationship between hearing conditions and work time of two subgroups (in the frequencies of 3, 4, 6 kHz). Scatter plots and linear fitting show the results of pure tone audiometry thresholds as work time increases.
Immunofluorescence staining
The gene encoding von Willebrand factor (VWF) showed the highest number of mutations in the current cohort. VWF is a macromolecular polysaccharide protein, associated with platelet adhesion and aggregation at the site of vascular injury [Citation11]. VWF had harmful mutations in 9 NIHL subjects and locus details are shown in . shows the Sanger sequencing validation of VWF gene mutation sites in NIHL patients in .
Figure 6. Sanger sequencing results for individuals carrying rare harmful mutations in VWF in the NIHL group.
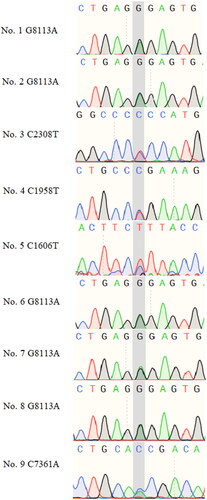
Table 5. Harmful mutation sites of VWF gene in NIHL population.
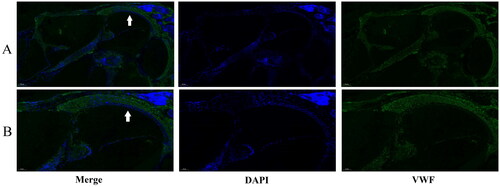
The distribution of VWF in the inner ear has not been previously determined. Immunofluorescence staining of cochlear sections showed VWF expression to be concentrated in the spiral ligament with a small amount in the stria vascularis ().
Figure 7. Immunofluorescence staining of VWF in c57BL/6 mouse cochlea. (A is 100×, B is 200×. The white arrows indicate the spiral ligament).
Discussion
The etiology of noise-induced hearing loss is complex and includes interaction of occupational with environmental noise exposure, drug use, vibration and genetic factors [Citation4]. Cochlear sensory cells and outer hair cells are damaged in NIHL and oxidative stress and excitatory synaptic toxicity are the main pathological mechanisms [Citation12]. Noise-induced hearing loss studies based on genetic changes have been performed in animal models and knockouts of SOD1 -/-, GPX1 -/-, PMCA -/- and CDH23 +/- shown to increase sensitivity to noise [Citation4]. Knockout studies have shown that some genetic defects destroy cochlear structure, increasing inner ear susceptibility to NIHL. Single nucleotide polymorphisms (SNP) have been identified from epidemiological studies of NIHL susceptibility genes in specific populations with first-generation sequencing at known or specific sites. Many SNP sites in NIHL susceptibility genes have been linked to NIHL, including those in SOD2, KCNQ1, PCDH15 and HSP70 [4]. Such SNP genotypes may make subjects more susceptible to NIHL. Whole-exome sequencing is a high-throughput sequencing method which allows the detection of hundreds of SNPs in a single array, allowing identification of novel NIHL susceptibility genes. Such approaches facilitate an exploration of the genetic basis of NIHL susceptibility, enabling protection measures and personalized treatment.
Mutations in the gene encoding von Willebrand factor (VWF) appeared in 9 NIHL individuals of the current cohort, the highest frequency of any NIHL susceptibility gene. Cluster analysis showed VWF mutations to be present in subgroups A and B. The A1 domain of VWF interacts with type I, II and IV collagen in the cochlea [Citation13]. Two mutations (c.3995G > T and c.5618C > T) of the D3-D4 domain of VWF have been previously linked to non-syndromic sensorineural hearing loss (NSHL) [Citation14]. Moreover, VWF has been implicated in sudden sensorineural hearing loss associated with thrombosis [Citation15]. In addition, two proteins which showed a high mutation rate in the current NIHL cohort, COL5A1 and MYO18A, may be related to cell adhesion. Mutation may reduce cochlear hair cell adhesion, leading to hair cell shedding [Citation16]. Adhesion-related gene COL5A1 have been previously linked to hearing loss and may cause changes in cochlear collagen expression and adhesion related mechanisms. Excessive movement of the cochlear basement membrane caused by excessive noise results in stretching of Corti organs and damage to intercellular connections and adhesion between cells and the extracellular matrix [Citation17]. VWF was found to be expressed in the spiral ligament, bone spiral plate, membrane spiral plate at the junction of hair cells and cochlea and blood vessels in our immunofluorescence staining results. Its concentration at these sites may be related to the stability of cochlear fine structure and microthrombosis. Further studies on animal models may allow the investigation of functions of novel gene mutations. In our study, only GJB4 has been studied in NIHL population susceptibility. Van et al. and Pawelczyk et al. found that SNP sites rs1998177 and rs755931 of the GJB4 gene are associated with NIHL risk. Other genes, such as the Myosin family and Collagen family genes, have only been studied in the pathogenesis of deafness, but no susceptibility has been found in NIHL. Therefore, we have provided a potentially novel NIHL susceptibility gene mutation profile.
Unsupervised machine learning algorithm clustering analysis allowed the identification of two subgroups, A and B, with differing characteristics of susceptibility. Subjects in subgroup A had more severe hearing loss than those in subgroup B following shorter noise exposure times. Subgroup A members had poorer hearing at 3 kHz and differences between subgroups were greater during longer work time. Differences may result from variations in NIHL susceptibility genes between the two groups. Subgroup A members are likely to show a more rapid deterioration of hearing with increased noise exposure. Supervised machine learning algorithms may be divided into regression-based methods, such as logistic regression, neural network and SVM, or tree-based methods, such as decision trees and random forests. Regression-based methods use a polynomial parameter or nonparametric regression to map the correlation between multidimensional input and output data [Citation18]. Tree-based methods usually use binary decision splitting rules to model the relationship between input and output data [Citation18]. MLP is an algorithm in a neural network which allows complex nonlinear problems to be randomly solved and which performs well in many other group analyses [Citation19]. The MLP algorithm showed better performance than other algorithms in predicting the classification of possible susceptibility genes during the current study and had 74.2% accuracy. The MLP prediction model had the highest AUC, accuracy and sensitivity and showed good discrimination and stability making it suitable for research and clinical use.
Conclusion
NIHL susceptibility genes were screened by whole-exome sequencing and mutations linked to the NIHL population identified. Genes were analyzed by machine learning algorithm to indicate a subgroup classification method with utility for predicting an individual’s susceptibility to noise exposure. The machine learning model allowed the identification of two genetic subgroups of the NIHL population. Four machine learning algorithms were used to predict and classify the NIHL clinical subgroups with good results for the test set. The expression pattern of VWF in mouse cochlea was shown. A WES-based method for gene screening and hearing loss prediction is introduced. Functional verification of the genes identified to clarify their roles in the occurrence and development of NIHL is planned.
Ethical approval
The authors are accounts for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). The study was approved by the ethical committee of PLA Strategic Support Force Characteristic Medical Center (No. K2019(03)) and informed consent was taken from all individual participants. All study subjects signed written informed consent.
Acknowledgements
The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated and analyzed during this study are included in this published article and its additional information files.
Additional information
Funding
References
- Royster JD. Preventing noise-induced hearing loss. N C Med J. 2017;78(2):113–117.
- Vlaming MSMG, MacKinnon RC, Jansen M, et al. Automated screening for high-frequency hearing loss. Ear Hear. 2014;35(6):667–679.
- Mao HY, Chen Y. Noise-Induced hearing loss: updates on molecular targets and potential interventions. Neural Plast. 2021;2021:1–16.
- Sliwinska-Kowalska M, Pawelczyk M. Contribution of genetic factors to noise-induced hearing loss: a human studies review. Mutat Res. 2013;752(1):61–65.
- Ngiam KY, Khor IW. Big data and machine learning algorithms for health-care delivery. Lancet Oncol. 2019;20(5):E262–e273.
- Henarejos-Castillo I, Aleman A, Martinez-Montoro B, et al. Machine learning-based approach highlights the use of a genomic variant profile for precision medicine in ovarian failure. JPM. 2021;11(7):609.
- do Valle IF, Giampieri E, Simonetti G, et al. Optimized pipeline of MuTect and GATK tools to improve the detection of somatic single nucleotide polymorphisms in whole-exome sequencing data. BMC Bioinformatics. 2016;17(Suppl 12):341.
- Kosub S. A note on the triangle inequality for the jaccard distance. Pattern Recogn Lett. 2019;120:36–38.
- Han H, Guo XL, Yu H. Variable selection using mean decrease accuracy and mean decrease gini based on random Forest, 2016 7th IEEE International Conference on Software Engineering and Service Science (ICSESS), Beijing, 2016, pp. 219-224, doi: 10.1109/ICSESS.2016.7883053
- AlKaabi LA, Ahmed LS, Al Attiyah MF, et al. Predicting hypertension using machine learning: findings from Qatar biobank study. PLOS One. 2020;15(10):e0240370.
- Zhang YX, Chen FW, Yang AZ, et al. The disulfide bond Cys2724-Cys2774 in the C-terminal cystine knot domain of von Willebrand factor is critical for its dimerization and secretion. Thrombosis J. 2021;19(1):94.
- Hong OS, Kerr MJ, Poling GL, et al. Understanding and preventing noise-induced hearing loss. Dis Mon. 2013;59(4):110–118.
- Nagy I, Trexler M, Patthy L. The second von Willebrand type a domain of Cochlin has high affinity for type I, type II and type IV collagens. FEBS Lett. 2008;582(29):4003–4007.
- Kim AR, Chang MY, Koo JW, et al. Novel TECTA mutations identified in stable sensorineural hearing loss and their clinical implications. Audiol Neurootol. 2015;20(1):17–25.
- Drouet L, Hautefort C, Vitaux H, et al. Plasma serotonin is elevated in adult patients with sudden sensorineural hearing loss. Thromb Haemost. 2020;120(9):1291–1299.
- Davis RR, Kozel P, Erway LC. Genetic influences in individual susceptibility to noise: a review. Noise Health. 2003;5(20):19–28.
- Cai QF, Patel M, Coling D, et al. Transcriptional changes in adhesion-related genes are site-specific during noise-induced cochlear pathogenesis. Neurobiol Dis. 2012;45(2):723–732.
- Mehta P, Bukov M, Wang CH, et al. A high-bias, low-variance introduction to machine learning for physicists. Phys Rep. 2019;810:1–124.
- Kaczmarek E, Jamzad A, Imtiaz T, et al. Multi-Omic graph transformers for cancer classification and interpretation. Pac Symp Biocomput. 2022;27:373–384.