
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Productions of mono-/di-cyclic aromatic hydrocarbons as well as smaller stable species from extremely fuel-rich CH4/air mixtures (equivalence ratio of 1.7–6.0 and fuel-to-mixture ratio of 15–38 mol.%) near sooting limit in terms of temperature, were investigated using a micro flow reactor with a controlled temperature profile at maximum wall temperature of 1300 K. Species measurements of O2, H2, CO, CO2, CH4, C2H2, C2H4, C2H6, benzene, toluene, styrene and naphthalene were performed with GC and GC/MS analysis. One-dimensional computations were also conducted with several detailed chemical kinetics. Most of the mechanisms comparably well predicted the smaller species except C2H2 (acetylene), which was overestimated by all the mechanism especially at moderate equivalence ratio (Ø3.0). There were large discrepancies between measured and computed mole fractions of benzene and naphthalene at high equivalence ratio (Ø
4.0). Reaction path analysis indicated that reaction pathway branched from C2H3 reacting with methyl radical, which competes with C2H2 production, showed relatively low contribution to benzene formation at moderate equivalence ratio. Therefore, improvements of chemical kinetics with further consideration of reactions with methyl radical are necessary for precise prediction of products where abundant amounts of methyl radical exist.
Introduction
Products from fuel-rich combustion have been widely investigated mainly from two practical aspects. One aspect is to understand precise composition of reformed gases in fuel reforming process. Reformed gas is utilized for various systems such as engines (Tartakovsky and Sheintuch Citation2018) and fuel cells (Milcarek et al. Citation2019). While much CO and H2 generation is expected with the increase of equivalence ratio, soot formation becomes a concern even at intermediate temperatures (around 1300 K) in the flame-assisted fuel cells due to its relatively long residence time. Therefore, it is important to know the reforming conditions to operate reformation systems with the best performance. Another aspect is to explicate the mechanisms of formation/growth of soot and its precursor aromatic species, which have harmful effects on human body and environment, while widely being used in industry as chemical materials. Many studies have been conducted at fuel-rich conditions to investigate sooting limits (Glassman Citation1988; Takahashi and Glassman Citation1984) and growth of soot and its precursors (Richter and Howard Citation2000; Wang Citation2011). These studies mainly focused on fuels containing two or more C-atoms. However, because the use of natural gas has been increasing, understanding of products from combustion of methane, which is a main component of natural gas, at fuel-rich conditions has become significantly important. As shown in , measurements of soot, aromatic species and smaller species from fuel-rich methane combustion were conducted by employing several methods, e.g. premixed flat-flame burners (Ahmed et al. Citation2016; Alfè et al. Citation2010; Castaldi, Vincitore, Senkan Citation1995; Li et al. Citation2012; Marinov et al. Citation1996; Melton, Vincitore, Senkan Citation1998; Xu and Faeth Citation2000), jet-stirred reactor (Cong and Dagaut Citation2008) and plug-flow reactors (Köhler et al. Citation2016; Skjøth-Rasmussen et al. Citation2004) near atmospheric pressure.
Figure 1. Experimental conditions of soot and species measurements for fuel-rich CH4 combustion near atmospheric pressure operated by various methods: burner stabilized flames (Ahmed et al. Citation2016; Alfè et al. Citation2010; Castaldi, Vincitore, Senkan Citation1995; Li et al. Citation2012; Marinov et al. Citation1996; Melton, Vincitore, Senkan Citation1998; Xu and Faeth Citation2000); jet-stirred reactor (Le Cong and Dagaut Citation2008); plug-flow reactors (Köhler et al. Citation2016; Skjøth-Rasmussen et al. Citation2004); and a micro flow reactor with a controlled temperature profile (this study).
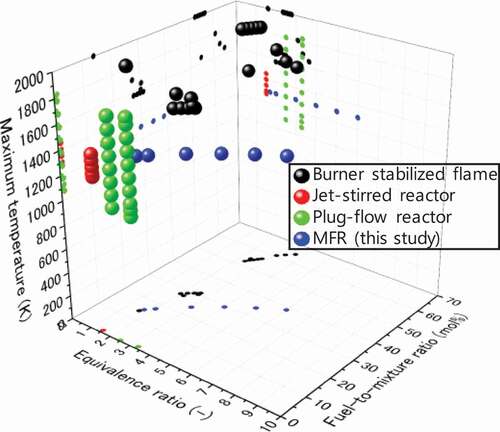
Modelling studies on growth of soot precursors such as polycyclic aromatic hydrocarbons (PAHs) in methane flame (Chernov et al. Citation2014; Jin et al. Citation2015; Marinov et al. Citation1996; Slavinskaya and Frank Citation2009) were also carried out. However, there is a gap in fuel-to-mixture ratio between the flame studies (approximately 27–65 mol.% at Ø2), which are close to practical conditions, and the reactor studies (less than 1 mol.%). This is because of the limitation of a self-sustained flame in the flame studies and large dilution to suppress temperature change in the reactor studies. As rate of formation of first aromatic ring is a primary factor to control soot formation (Glassman Citation1988), investigation of gas-phase species at both of high equivalence ratio and high fuel-to-mixture ratio conditions is necessary for further understanding of products from fuel-rich methane combustion. Particularly, it is important to gain new insight into the formation of the first aromatic ring under conditions where significant amount of methyl radical exists, as well as PAH growth with such as methyl addition/cyclization (Shukla, Miyoshi, Koshi Citation2010). To realize the extremely fuel-rich conditions, i.e. high equivalence ratio and high fuel-to-mixture ratio, a micro flow reactor with a controlled temperature profile (Maruta et al. Citation2005) (MFR) was employed in this study.
MFR comprises a quartz tube whose inner diameter is smaller than ordinary quenching diameter, and an external heat source which gives a stationary temperature profile along the inner surface of the reactor, as show in the upper part of . Heated by the external heat source, MFR is capable of handling combustion of mixtures even outside of the flammability limits. In our previous studies on near sooting-limit flames using MFR (Dubey et al. Citation2016; Nakamura et al. Citation2014), four types of flame and soot responses were observed depending on equivalence ratio and inlet flow velocity: only flame; flame with soot; only soot; and none of them. For the second case, flame and sooting regions were spatially separated in the flow direction, as shown in .
Figure 2. Schematic and direct image (Dubey et al. Citation2016) of flame and soot observation in MFR.
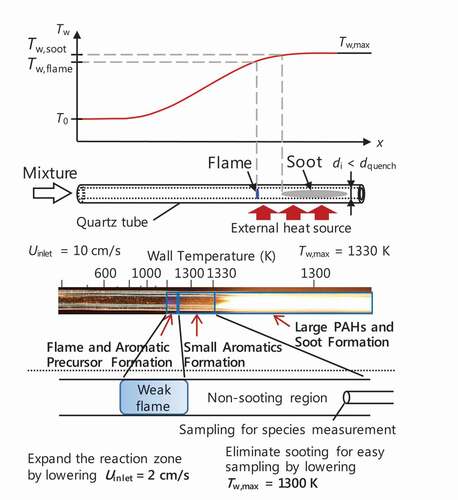
Two types of flame regime exist at fuel-rich conditions depending on inlet flow velocity: weak flame in low and normal flame in high inlet flow velocity conditions. At low inlet flow velocity (weak flame) conditions, gas temperature is strongly governed by wall temperature because of small heat release from the reaction zone and also small Peclet number which is the nature of MFR. Consequently, a rapid increase in gas temperature is suppressed resulting in no need of high dilution of mixtures. From these characteristics of MFR at low inlet flow velocity conditions, the extremely fuel-rich conditions can be realized and a series of phenomena up to a certain temperature range, i.e. only gas-phase reactions even near sooting limit, can be extracted by controlling maximum wall temperature (Tw,max). In our previous studies applying this method, species measurements of PAHs for fuel-rich acetylene (Nakamura et al. Citation2014) and n-/iso-cetane (Nakamura et al. Citation2015) were performed. Flame positions and species measurement of C1–C2 species for fuel-rich methane/air at Tw,max = 1200 K were also investigated (Dubey et al. Citation2019). In addition to the production of the smaller species reported in (Dubey et al. Citation2019), it is important to know that of aromatic species where the production becomes significant as well as near sooting limit in terms of temperature, as shown in .
Figure 3. Benzene production as a function of maximum wall temperature at Ø = 6.0 in MFR. See following sections for experimental and computational methods.
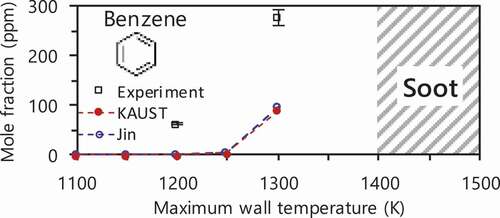
In this study, species measurements of C0–C2 species and aromatic hydrocarbons for extremely fuel-rich methane/air mixtures at Tw,max = 1300 K, where near sooting limit and production of aromatic species drastically starts increasing as shown in , were performed by GC and GC/MS connected to MFR. One-dimensional computations were also performed with several chemical kinetics to investigate the species productions.
Experimental method
shows a schematic of experimental setup. A quartz tube with an inner diameter of 2 mm was used as a reactor channel.
Figure 4. Schematic of experimental setup. The temperature profile shown in the figure was measured in experiments and used in computations.
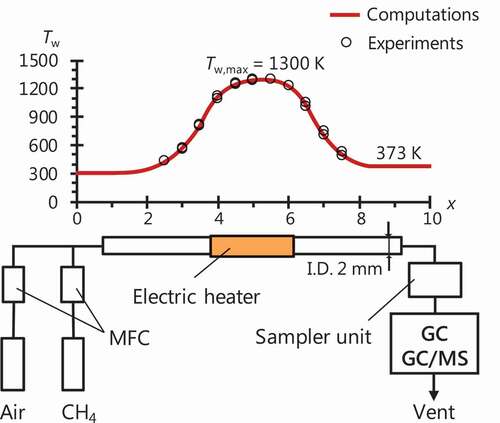
CH4/air mixtures were supplied to the reactor at a mean inlet flow velocity of 2 cm/s. Equivalence ratio (Ø) was varied over a range of 1.7–6.0 (fuel-to-mixture ratio of 15–38 mol.%) by controlling flow rates of fuel and air with mass flow controllers. Experiments were conducted at atmospheric pressure. From our previous studies for CH4/air mixtures in MFR (Dubey et al. Citation2016; Nakamura et al. Citation2014), soot formation was observed at Tw,max = 1300 K with Tw,max-region of approximately 4-cm-long heated by a hydrogen/air flat-flame burner as an external heat source. In this study, an electric heater was attached to the reactor as the heat source aiming to shorten the length of Tw,max-region, which was approximately 0.5 cm long. Consequently, soot formation was not observed at Tw,max = 1300 K but clearly observed at Tw,max = 1400 K. Therefore, a stationary temperature profile with the range of 300–1300 K was provided to focus on gas-phase products near sooting limit. The temperature profile of the inner wall surface of the reactor was measured with a K-type thermocouple inserted from the downstream of the reactor. Exhaust gas was directly introduced into a sampler unit from the exit of MFR through a sampling line. The line was heated as 373 K to prevent water condensation. Sampled gas was then analysed by GC (Shimadzu GC-2010 Plus) or GC/MS (Agilent 7890/5975).
O2, H2, CO, CO2, CH4, C2H2, C2H4 and C2H6 were measured by GC. For GC analysis, the sampling volume was set to 50 μl and a coupled two MICROPACKED-ST (3.0 m × 1.0 mm I.D.) capillary column was employed. A barrier discharge ionization detector (BID) was adopted as a detector and its temperature was set to 483 K. Standard gases were used for the quantification of the species.
Benzene, toluene, styrene and naphthalene were measured by GC/MS. For GC/MS analysis, the sampling volume was set to 500 μl and DB-5 ms Ultra Inert (60 m × 0.25 mm I.D.) was employed as a column. Temperatures of the ionization source and quadrupole were set to 513 K and 439 K, respectively. Note that for GC/MS experiment, 21%O2/1%Ar/N2 synthetic air was used as “air”. Internal standard method was applied for calibration of benzene using Ar as an internal standard substance along with standard gas of benzene. Relative response factors were applied for quantification of toluene, styrene and naphthalene. In addition to the measured species, cyclopentadiene, ethylbenzene, xylene isomers, indene and methylnaphthalene were identified but their signals were too small to be quantitatively discussed. Helium was used as a carrier gas for both of GC/BID and GC/MS experiments. Experiments were repeated at least three times for each condition and uncertainties in measured mole fractions were shown as error bars.
Computational method
To validate various chemical kinetic models and to conduct analysis of productions of the measured species, computations were performed by modelling the MFR system. The flow field in MFR can be assumed as reactive flow without boundary layer at low inlet flow velocity conditions. The usage of one-dimensional computations for MFR was justified by comparison of one- and two-dimensional computations (Grajetzki et al. Citation2019). Therefore, one-dimensional steady-state computations were conducted using PREMIX code in ANSYS Chemkin-Pro v19.0 with an additional term of convective heat transfer between the gas and the reactor wall in a gas-phase energy equation (Maruta et al. Citation2005). Computational conditions (mixture compositions, inlet flow velocity, wall temperature profile () and pressure) were the same as those in the experiment. Computational domain was set to 0–10 cm. Species mole fractions at x = 10 were compared with experimental results. Computed species profiles of the aromatic species in MFR are shown in Supplementary Fig. S1. Previous studies (Kizaki et al. Citation2015; Saiki and Suzuki Citation2013) reported that radical quenching effect on a quartz surface in the heated narrow channel under atmospheric pressure is negligibly small. Therefore, any surface reaction mechanism for radical quenching effect was not considered in this study. As detailed chemical kinetic models, KAUST-Aramco PAH Mech 1.0 (Selvaraj et al. Citation2016) (397 species, 2346 reactions, includes up to C24; hereafter called as KAUST), Jin mechanism (Jin et al. Citation2017) (238 species, 1689 reactions, includes up to C18; Jin) and PRF + PAH mechanism (Version 1412, December 2014) (CRECK Modeling Group, Citation2014) (176 species, 6067 reactions, includes up to C20; CRECK) were used.
Results and discussion
Results of species measurement and computation
shows experimental and computational results of mole fractions of O2, H2, CO, CO2, CH4, C2H2, C2H4, C2H6, benzene, toluene, styrene and naphthalene. Results are stated in three parts: small species up to C1 species; C2 hydrocarbons; and mono-/di-cyclic aromatic hydrocarbons.
Figure 5. Measured (open square) and computed (circle with line) mole fractions of O2, H2, C1–C2 and aromatic species for CH4/air mixtures at Ø = 1.7–6.0 (fuel-to-mixture ratio of 15–38 mol.%) and Tw,max = 1300 K.
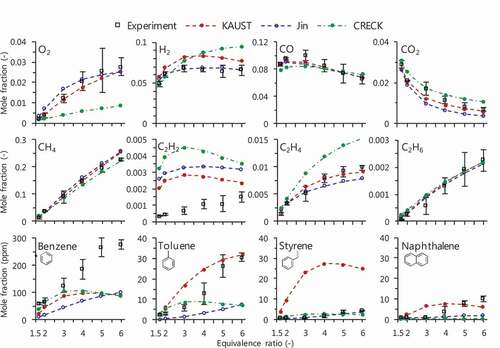
First, results for O2, H2 and C1 species are examined. The measured O2 mole fraction increases as equivalence ratio increases. This tendency is caused by a suppression of reactions that highly contribute to consumption of O2, e.g. CO and CO2 production in C1 reaction pathway, competing with proceeding of reaction pathway in higher hydrocarbons. KAUST and Jin quantitatively predict the measured O2 mole fraction, while CRECK underestimates O2 mole fraction especially at high equivalence ratio. The measured H2 mole fraction increases as equivalence ratio increases until Ø = 3.0, and then shows almost a constant value at higher equivalence ratio. KAUST and Jin qualitatively reproduce the measured H2 mole fraction, while CRECK shows increasing trend. The measured CO mole fraction increases at moderate equivalence ratio and decreases at high equivalence ratio as equivalence ratio increases, having a peak at around Ø = 3.0. The measured CO2 mole fraction decreases as equivalence ratio increases. All the mechanisms predict the measured CO and CO2 mole fractions well. The measured CH4 mole fraction shows a linear increase with increasing equivalence ratio. The computational CH4 mole fraction with CRECK agrees with the experimental result, while KAUST and Jin slightly overestimate CH4 mole fraction at high equivalence ratio. For these O2, H2 and C1 species, which show larger mole fractions among the measured species, the mechanisms reproduce the experimental results comparably well.
Secondly, results for C2 hydrocarbons are examined. Measured mole fraction of C2H2, one of the most important aromatic/soot precursors, monotonically increases as equivalence ratio increases. However, none of the mechanisms predicts this trend especially at high equivalence ratio: KAUST and CRECK have a peak at Ø = 3.0 and then tend to decrease; Jin shows almost a constant value at high equivalence ratio. It is notable that all the mechanisms overestimate C2H2 mole fraction over the equivalence ratio. This overestimation of C2H2 will be discussed in Section 4.2. The measured C2H4 mole fraction marks the largest value among C2 hydrocarbons, but its increasing trend becomes gradually small as equivalence ratio increases. KAUST quantitatively reproduces the measured C2H4 mole fraction. Although Jin slightly underestimates C2H4 mole fraction at high equivalence ratio, it agrees with the experimental result at moderate equivalence ratio. CRECK shows larger value than measured C2H4 mole fraction by a factor of more than 1.5 at high equivalence ratio. The measured C2H6 mole fraction shows a linear increase with increasing equivalence ratio. All the mechanisms quantitatively predict C2H6 mole fraction. As a brief summary for prediction of C2 hydrocarbons, model dependence in C2H2 mole fraction is significant compared to C2H4 and C2H6 mole fractions.
Finally, mono-/di-cyclic aromatic hydrocarbons such as benzene, toluene, styrene and naphthalene were quantitated at Tw,max = 1300 K. The measured benzene mole fraction increases as equivalence ratio increases. KAUST and CRECK agree with the experimental result at moderate equivalence ratio (Ø3.0), while they show almost constant values or even a slightly decreasing trend at high equivalence ratio (Ø
3.0). Consequently, discrepancy between the measured and computed benzene mole fractions becomes large as equivalence ratio increases. In contrast, Jin qualitatively reproduces the upward trend in measured benzene mole fraction over the equivalence ratio. However, it underestimates benzene mole fraction by a factor of 3. Measured toluene and styrene mole fractions also increase as equivalence ratio increases. KAUST predicts toluene mole fraction well although it overestimates styrene mole fraction. Jin qualitatively predicts toluene mole fraction even though it slightly underestimates the mole fraction, and agrees with the measured styrene mole fraction. Comparing the computed toluene and styrene mole fractions of KAUST with those of Jin, the latter mechanism shows smaller value than the former one. For both toluene and styrene mole fractions, CRECK shows similar trend as is obtained in prediction of benzene mole fraction, i.e. decreasing trend at high equivalence ratio. A drastic increase in measured naphthalene mole fraction is observed at around Ø = 4.0, which none of the mechanisms predicts the trend; KAUST has a peak at Ø = 4.0 followed by decreasing trend at higher equivalence ratio; Jin monotonically increases over the equivalence ratio; and CRECK shows approximately an order of the magnitude smaller than the measured naphthalene mole fraction. For these mono-/di-cyclic aromatic hydrocarbons, it is found that improvements of reaction mechanisms are required especially for production of benzene at Ø
4.0.
From these comparisons, significant discrepancies between measured and computed mole fractions were observed for C2H2 and benzene, which show relatively large mole fractions and are major aromatic/soot precursors. Therefore, discussion focusing on these species will be conducted in the following sections.
Overestimation of C2H2 mole fraction
All the mechanisms overestimated C2H2 mole fraction as mentioned in the previous section. shows computational results of C2H2 mole fraction performed with various mechanisms including four additional chemical mechanisms (Metcalfe et al. Citation2013; San Diego Mechanism, Citation2016; Smith, Tao, Wang Citation2016; Yang et al. Citation2017), and the experimental result.
Figure 6. C2H2 prediction performed by various chemical kinetics: KAUST (Selvaraj et al. Citation2016), Jin (Jin et al. Citation2017) and CRECK (CRECK Modeling Group, Politecnico di Milano Citation2014) as mechanisms including PAH growth; AramcoMech 1.3 (Metcalfe et al. Citation2013), San Diego mechanism (Citation2016) (San Diego Mechanism web page, University of California at San Diego Citation2016), HP-Mech (Yang et al. Citation2017) and FFCM-1 (Smith, Tao, Wang Citation2016) as non-PAH mechanisms. Black long broken line shows the mole fraction of measured benzene by a factor of 3.
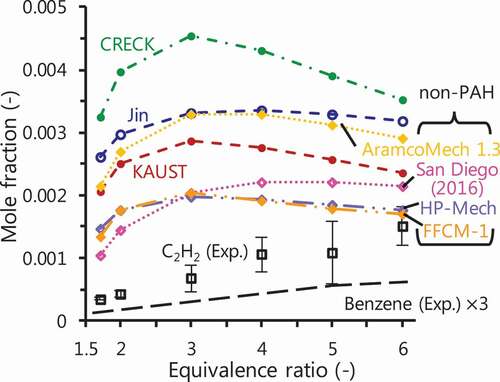
Computational results of the smaller species with the additional mechanisms are shown in Supplementary Fig. S2. The additional mechanisms do not include PAH formation, but are recently updated. Even these new mechanisms, however, overestimate C2H2 mole fraction. The overestimation of C2H2 is also reported in flame (Li et al. Citation2012; Xu and Faeth Citation2000), plug-flow reactor (Köhler et al. Citation2016) and shock tube (Herzler et al. Citation2019) studies for methane fuel. Most of the computational results with the additional (non-PAH) mechanisms show smaller values than those of the PAH mechanisms. However, even considering production of benzene, which possibly shows the largest amount among aromatic hydrocarbons in this situation, all the mechanisms still overestimate C2H2 mole fraction especially at moderate equivalence ratio (Ø2.0). This is also obvious from comparison of the computational results with AramcoMech 1.3 (non-PAH) and KAUST, which is based on AramcoMech 1.3. Considering PAH growth decreases the mole fraction of C2H2 at high equivalence ratio (Ø
2.0) and the reduced amount is in similar magnitude to the threefold value of benzene mole fraction computed with KAUST. Although the PAH sub-set contributes to improving the discrepancy at high equivalence ratio, little effect is observed on prediction of C2H2 mole fraction at moderate equivalence ratio. Therefore, this discrepancy is considered to be mainly caused by insufficient modelling in lower hydrocarbon chemistry more than that in growth of aromatic species.
shows results of sensitivity analysis performed for C2H2 mole fraction at equivalence ratios of 1.7, 4.0 and 6.0 with Jin and HP-Mech, which shows relatively small C2H2 mole fraction among the mechanisms. Top 10 sensitivity coefficients were taken from locations at maximum rate for C2H2 production. Reactions with indexes of Rx are presented here for discussion.
Figure 7. First order, A-factor sensitivity coefficients for C2H2 mole fraction at Ø = 1.7, 4.0 and 6.0 performed with (a) Jin and (b) HP-Mech. Top 10 sensitive reactions at location of maximum rate of C2H2 production for each condition are shown.
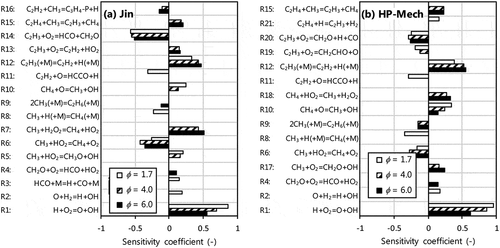
For the both mechanisms, hydrogen-oxygen reactions (R1, R2), initial-stage reactions of methane (R6, R8, R10) and reactions involving C2H2 (R11, R12) show high sensitivity coefficients of C2H2 mole fraction at moderate equivalence ratio. Hydrogen-oxygen reactions such as R1 (H+O2=O +OH) and R2 (O +H2=H +OH) highly affect to other combustion characteristics as well, e.g. laminar burning velocity (See Supplementary Fig. S3 for sensitivity coefficients of the laminar burning velocity of methane at varied equivalence ratios performed with Jin and HP-Mech). R8 (CH3+H (+M)=CH4 + (M)) was also sensitive to the laminar burning velocity in the both mechanisms as well. Therefore, it seems to be strict to improve the model prediction of C2H2 by modifying these reactions. R6 (CH3 +HO2=CH4 +O2) shows a negative sensitivity to C2H2 mole fraction. The rate constants for R6 obtained from experimental (Hong et al. Citation2012; Scire, Yetter, Dryer Citation2001) and theoretical (Jasper, Klippenstein, Harding Citation2009; Mai et al. Citation2014; Zhu and Lin Citation2001) studies included relatively large uncertainty (by a factor of ~10 in the former and ~5 in the latter) (Onda et al. Citation2019). Slightly higher rate constant than that used in AramcoMech 1.3, HP-Mech, FFCM-1 and Jin was suggested in (Onda et al. Citation2019), and the suggested rate constant seems reasonable for the result of sensitivity analysis in this study. R10 (CH4+O=CH3 +OH) has been studied well (Baulch et al. Citation2005) and difference of the rate constants used in the mechanisms was within a factor of approximately 1.2. R11 (C2H2+O=HCCO + H) is a main reaction of C2H2 consumption competing with C2H2+O=CH2 +CO. Branching ratio of the two reactions was suggested as approximately 0.8:0.2 with little dependence on temperature (Baulch et al. Citation2005; Nguyen, Vereecken, Peeters Citation2006). Similar branching ratios (0.8:0.2) and rate constants (within a factor of approximately 1.2) were used in the most mechanisms except CRECK and San Diego (Citation2016). CRECK used much lower rate constants of the both reactions, therefore, these lower rate constants are considered as one of the factors that led to the significant overestimation of C2H2 mole fraction compared to the other mechanisms. However, a rate constant of R11 used in San Diego Mechanism web page, University of California at San Diego (Citation2016) was also lower than that used in the other mechanisms at low to intermediate temperatures. Therefore, in addition to reactions that contribute to consumption of C2H2, reactions that suppress/compete with production of C2H2 need to be examined further. R12 (C2H3 (+M)=C2H2+H (+M)) was one of those reactions that highly contributes to C2H2 production under the current conditions. The high- and low-pressure limit rate constants for R12 reported by (Knyazev and Slagle Citation1996; Miller and Klippenstein Citation2004) were in fairly good agreement and were used in the most of the mechanisms. However, other major pathways of vinyl such as reactions of C2H3 +O2, which compete with the decomposition reaction leading to C2H2, were varied depending on mechanisms. The reactions of C2H3 +O2 included in each mechanism are shown in .
Table 1. Reactions of C2H3 +O2=products included in the mechanisms
The main products of C2H3 +O2 were considered as HCO + CH2O (R14) at lower temperatures and CH2CHO + O (R19) at higher temperatures (Curran Citation2019). In addition to these channels, C2H3 +O2=C2H2 +HO2 (R13) was considered as the third significant channel in most of the mechanisms. Although most of the mechanisms include those reactions, it was indicated that the collisional stabilization of C2H3O2 competes with R14 and R19 at higher pressure (>10 atm) and that OCHCHO + H and CHCHO + OH (not included in any of the mechanisms) were more significant product channels than R13 (Goldsmith et al. Citation2015). The pressure-dependent rate constants for C2H3 +O2 reactions are considered only in HP-Mech and better if they are considered in the other mechanisms as well. In addition to these reactions, recently studied reactions in C2H2 chemistry (e.g. C2H2 +HO2 reactions (Gimenez-Lopez et al. Citation2016)) could be candidates to be updated in the mechanisms. Therefore, it is indicated that reactions related to C1–C2 species need to be further investigated and that improvements of chemical kinetics in lower hydrocarbon chemistry are required for accurate prediction of C2H2 especially at moderate equivalence ratio.
Formation of benzene
To investigate formation of benzene for fuel-rich combustion of CH4, reaction path analysis was performed with KAUST and Jin as shown in . Main reaction pathway from CH4 to C2H4 is not shown in the figure because it was identical for both mechanisms. There are mainly three benzene formation pathways: (P1) C4Hx + C2Hy reactions; (P2) C3Hx + C3Hy reactions; (P3) cyclic species reactions. As examined in Section 4.1, KAUST quantitatively reproduced the measured benzene mole fraction at moderate equivalence ratio (Ø3.0), while Jin reproduced the overall upward trend in benzene mole fraction even at high equivalence ratio (Ø
4.0). Formation of benzene will be discussed at two typical cases: Ø = 2.0 for the moderate equivalence ratio case; and Ø = 6.0 for the high equivalence ratio case.
Figure 8. Reaction pathways of benzene formation analysed with (a) KAUST and (b) Jin at a maximum rate of benzene production, and Ø = 2.0 and 6.0. Numbers in parenthesis are percent contribution to production of benzene, toluene and styrene. The measured species are shown in boxes.
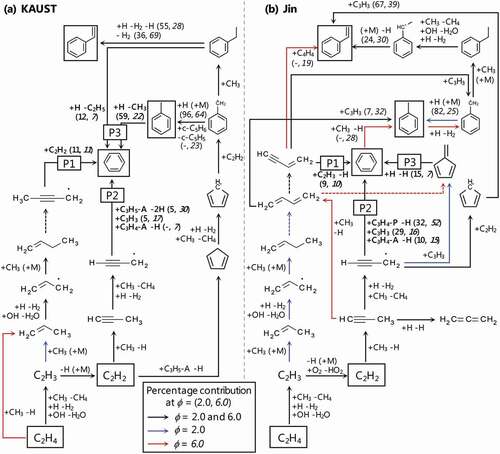
First, in at Ø = 2.0, P1 branches from C2H3 reacting with methyl radical. This is an important reaction proceeding to C3 pathway under large amounts of methyl radical exist (Fahr, Laufer, Tardy Citation1999). The pathway then proceeds up to C4 species, which produces benzene by C2H2 addition. P2 proceeds through propyne (C3H4-P) and propargyl (C3H3), which are initiated from C2H2 reacting with methyl radical, followed by benzene formation. The reaction pathway of C3 species is known to be significant for benzene formation (Miller and Melius Citation1992). However, contributions of these P1 and P2 to benzene formation show only 11% and 10% respectively at moderate equivalence ratio. For benzene formation of KAUST at this condition, P3 is dominant (71% of contribution in total). Particularly, reaction pathway which proceeds via toluene marks the highest contribution (59%). P3 proceeds via cyclopentadienyl (c-C5H5) and benzyl (C6H5CH2), which play important roles in formation/growth of aromatic species reacting with methyl radicals (Moskaleva, Mebel, Lin Citation1996; Shukla, Miyoshi, Koshi Citation2010; Slavinskaya and Frank Citation2009). In contrast, a dominant reaction pathway for benzene formation in at Ø = 2.0 is P2, which shows 71% of contribution. While P3 appears in Jin as well, it proceeds via fulvene, which is different from the one in KAUST, and shows only 15%. This difference is mainly because c-C5H5 +C2H2=C6H5CH2 (R22) is not included in Jin, resulting in smaller mole fractions of benzene and toluene than those in KAUST, as mentioned in Section 4.1 ().
When R22 was added to Jin as a trial, computational result showed larger mole fractions of the aromatic species than those performed by the original mechanism, and quantitatively reproduced measured benzene mole fraction at moderate equivalence ratio (Supplementary Fig. S4). This was because, as can be seen in , enhancing reaction pathway via benzyl promotes production of toluene and ethylbenzene (C6H5C2H5), which produces styrene, as well as benzene. As a result, the computed toluene mole fraction showed closer agreement with the measured mole fraction, while the computed styrene mole fraction slightly overestimated the measured mole fraction. There are, however, other possible pathways for reaction of c-C5H5 +C2H2 such as subsequence of indene formation through cycloheptatrienyl radical (Fascella et al. Citation2005), which is not included in the either mechanism. Therefore, close investigation is required to confirm the feasibility of the rate constant of R22 but it was implied that whether including R1 highly affects benzene formation via toluene at moderate equivalence ratio from this trial computation. Considering the overestimation of C2H2 at moderate equivalence ratio, it is suggested that improvements of chemical kinetics are required for reactions relevant to P1, which relatively less contributes to benzene formation in this situation, such as C2H3 +CH3 reaction (Richter and Howard Citation2000) as it competes with C2H2 production where abundant amounts of methyl radical exist.
Secondly, for the cases of Ø = 6.0, it is found that P2 is the dominant pathway of benzene formation for both KAUST and Jin. While P2 in Jin shows 83% of contribution to benzene formation, that in KAUST shows 54%. Even considering relatively high contribution of P3 in KAUST, the proportion of P2 in KAUST is still smaller than that in Jin. This is mainly because C3H3 +C3H4-P=benzene + H (R23), which is the most dominant reaction for benzene formation in Jin, is not included in KAUST. Therefore, R23 is considered as one of the factors that caused the different tendency in computed benzene mole fraction between Jin and KAUST at high equivalence ratio mentioned in Section 4.1. Consequently, the discrepancy between measured and computed benzene mole fraction for KAUST is enlarged at high equivalence ratio.
To examine the effect of R23 on benzene mole fraction at high equivalence ratio, a trial computation was conducted by adding R23 to KAUST (Supplementary Fig. S5). It was figured out that adding R23 increased the computed benzene mole fraction but it was still smaller than the measured value by a factor of 1.6, and that detailed improvements of chemical kinetics are required to sufficiently reproduce the measured benzene mole fraction. It should be also noted that there was no significant influence on computed mole fractions of the other measured species by adding the reaction.
Large amounts of CH3 exist under the current conditions as shown in .
Figure 9. Profiles of typical radicals for CH4/air mixtures at Ø = 6.0 computed with KAUST (red lines) and Jin (blue lines).
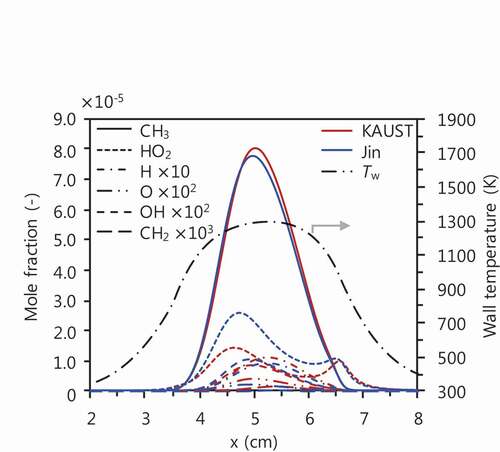
Although radicals such as CH2 and C2H3 could also be a candidate that enhance the growth of aromatic hydrocarbons (Liu et al. Citation2015; Shukla and Koshi Citation2012), reactions with CH3 would be significantly of importance in benzene formation from the combustion of fuel-rich methane. One possible pathway to benzene formation, which does not appear in , is subsequence of c-C5H5 +CH3 reaction leading to fulvene (Moskaleva, Mebel, Lin Citation1996; Sharma and Green Citation2009). Jin shows importance in the benzene formation from fulvene: fulvene + H=benzene + H (Jasper and Hansen Citation2013) (R24), which would be due to high H/C ratio in methane combustion, whereas fulvene formation is included only from C2 to C4 pathways. In contrast, KAUST includes the fulvene production channel from c-C5H5 +CH3 reaction but does not include benzene formation with R24. Therefore, improvements of chemical kinetics are necessary for better prediction of especially benzene mole fraction at extremely methane-rich conditions, where potentially abundant amounts of methyl radical exist, with further consideration of their impacts on formation/growth of the cyclic species.
Conclusions
Species measurements for extremely fuel-rich CH4/air mixtures near sooting limit conditions, which were equivalence ratio of 1.7–6.0 (fuel-to-mixture ratio of 15–38 mol.%) and maximum wall temperature of 1300 K, were conducted by using a micro flow reactor with a controlled temperature profile combined with GC/BID and GC/MS. KAUST-Aramco PAH Mech 1.0, Jin mechanism and CRECK mechanism were validated with mole fractions of O2, H2, CO, CO2, CH4, C2H2, C2H4, C2H6, benzene, toluene, styrene and naphthalene. Computational results of KAUST-Aramco PAH Mech 1.0 and Jin mechanism reproduced mole fractions of most of the measured smaller species, i.e. O2, H2 and C1–C2 species comparably well, while all the models overestimated mole fraction of C2H2. One- and two-ringed aromatic hydrocarbons (benzene, toluene, styrene and naphthalene) were quantitated in the experiment and mole fractions of those species were found to increase as equivalence ratio increased. KAUST-Aramco PAH Mech 1.0 agreed with measured benzene mole fraction at moderate equivalence ratio (Ø3.0), while it was not able to reproduce the upward trend observed in the experiment at high equivalence ratio (Ø
4.0). Although Jin mechanism underestimated benzene mole fraction over the equivalence ratio, it qualitatively reproduced the experimental result. Reaction path analysis of benzene formation performed for the two mechanisms indicated that a reaction pathway via toluene and a reaction of propargyl + propyne significantly affect benzene formation at respectively moderate and high equivalence ratios. There are, however, discrepancies between experimental and computational results of especially benzene as well as C2H2. Therefore, improvements of chemical kinetics with greater attention to methyl radical reactions related to production of C3 species and production/growth of cyclic species are necessary.
Declaration of conflicting interests
The authors declare no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Supplemental_Table_1.xlsx
Download ()Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Ahmed, A. M., S. Mancarella, P. Desgroux, L. Gasnot, J. F. Pauwels, and A. E. Bakali. 2016. Experimental and numerical study on rich methane/hydrogen/air laminar premixed flames at atmospheric pressure: Effect of hydrogen addition to fuel on soot gaseous precursors. Int. J. Hydrogen Energy 41 (16):6929. doi:https://doi.org/10.1016/j.ijhydene.2015.11.148.
- Alfè, M., B. Apicella, J. N. Rouzaud, A. Tregrossi, and A. Ciajolo. 2010. The effect of temperature on soot properties in premixed methane flames. Combust. Flame 157 (10):1959. doi:https://doi.org/10.1016/j.combustflame.2010.02.007.
- Baulch, D. L., C. T. Bowman, C. J. Cobos, R. A. Cox, T. Just, J. A. Kerr, M. J. Pilling, D. Stocker, J. Troe, W. Tsang, et al. 2005. Evaluated kinetic data for combustion modeling: Supplement II. J. Phys. Chem. Ref. Data 34 (3):757. doi:https://doi.org/10.1063/1.1748524.
- Castaldi, M. J., A. M. Vincitore, and S. M. Senkan. 1995. Micro-structures of premixed hydrocarbon flames: Methane. Combust. Sci. Technol. 107 (1–3):1. doi:https://doi.org/10.1080/00102209508907792.
- Chernov, V., M. J. Thomson, S. B. Dworkin, N. A. Slavinskaya, and U. Riedel. 2014. Soot formation with C1 and C2 fuels using an improved chemical mechanism for PAH growth. Combust. Flame 161 (2):592. doi:https://doi.org/10.1016/j.combustflame.2013.09.017.
- Cong, T. L., and P. Dagaut. 2008. Experimental and detailed kinetic modeling of the oxidation of methane and methane/syngas mixtures and effect of carbon dioxide addition. Combust. Sci. Technol. 180 (10–11):2046. doi:https://doi.org/10.1080/00102200802265929.
- CRECK Modeling Group, Politecnico di Milano. 2014. Primary Reference Fuels (PRF) + PAH mechanism. http://creckmodeling.chem.polimi.it
- Curran, H. J. 2019. Developing detailed chemical kinetic mechanisms for fuel combustion. Proc Combust Inst 37 (1):57. doi:https://doi.org/10.1016/j.proci.2018.06.054.
- Dubey, A. K., T. Tezuka, S. Hasegawa, H. Nakamura, and K. Maruta. 2016. Study on sooting behavior of premixed C1–C4 n-alkanes/air flames using a micro flow reactor with a controlled temperature profile. Combust. Flame 174:100. doi:https://doi.org/10.1016/j.combustflame.2016.09.007.
- Dubey, A. K., T. Tezuka, S. Hasegawa, H. Nakamura, and K. Maruta. 2019. Analysis of kinetic models for rich to ultra-rich premixed CH4/air weak flames using a micro flow reactor with a controlled temperature profile. Combust. Flame 206:68. doi:https://doi.org/10.1016/j.combustflame.2019.04.041.
- Fahr, A., A. H. Laufer, and D. C. Tardy. 1999. Pressure effect on CH3 and C2H3 cross-radical reactions. J Phys Chem A 103 (42):8433. doi:https://doi.org/10.1021/jp9923522.
- Fascella, S., C. Cavallotti, R. Rota, and S. Carrà. 2005. The peculiar kinetics of the reaction between acetylene and the cyclopentadienyl radical. J. Phys. Chem. A 109 (33):7546. doi:https://doi.org/10.1021/jp051508x.
- Gimenez-Lopez, J., C. T. Rasmussen, H. Hashemi, M. U. Alzueta, Y. Gao, P. Mar-shall, C. F. Goldsmith, and P. Glarborg. 2016. Experimental and kinetic modeling of C2H2 oxidation at high pressure. Int J Chem Kinet 48 (11):724. doi:https://doi.org/10.1002/kin.21028.
- Glassman, I. 1988. Soot formation in combustion processes. Proc Combust Inst 22 (1):295. doi:https://doi.org/10.1016/S0082-0784(89)80036-0.
- Goldsmith, F., L. B. Harding, Y. Georgievskii, J. A. Miller, and S. J. Klippenstein. 2015. Temperature and pressure-dependent rate coefficients for the reaction of vinyl radical with molecular oxygen. J. Phys. Chem. A 119 (28):7766. doi:https://doi.org/10.1021/acs.jpca.5b01088.
- Grajetzki, P., T. Onda, H. Nakamura, T. Tezuka, and K. Maruta. 2019. Investigation of the chemical and dilution effects of major EGR constituents on the reactivity of PRF by weak flames in a micro flow reactor with a controlled temperature profile. Combust. Flame 209:13. doi:https://doi.org/10.1016/j.combustflame.2019.06.021.
- Herzler, J., Y. Sakai, M. Fikri, and C. Schulz. 2019. Shock-tube study of the ignition and product formation of fuel-rich CH4/air and CH4/additive/air mixtures at high pressure. Proc Combust Inst 37 (4):5705. doi:https://doi.org/10.1016/j.proci.2018.05.120.
- Hong, Z., D. F. Davidson, K. Y. Lam, and R. K. Hanson. 2012. A shock tube study of the rate constants of HO2 and CH3 reactions. Combust. Flame 159 (10):3007. doi:https://doi.org/10.1016/j.combustflame.2012.04.009.
- Jasper, A. W., and N. Hansen. 2013. Hydrogen-assisted isomerizations of fulvene to benzene and of larger cyclic aromatic hydrocarbons. Proc Combust Inst 34 (1):279. doi:https://doi.org/10.1016/j.proci.2012.06.165.
- Jasper, A. W., S. J. Klippenstein, and L. B. Harding. 2009. Theoretical rate coefficients for the reaction of methyl radical with hydroperoxyl radical and for methylhydroperoxide decomposition. Proc. Combust. Inst. 32 (1):279. doi:https://doi.org/10.1016/j.proci.2008.05.036.
- Jin, H., A. Frassoldati, Y. Wang, X. Zhang, M. Zeng, Y. Li, F. Qi, A. Cuoci, and T. Faravelli. 2015. Kinetic modeling study of benzene and PAH formation in laminar methane flames. Combust. Flame 162 (5):1692. doi:https://doi.org/10.1016/j.combustflame.2014.11.031.
- Jin, H., G. Wang, Y. Wang, X. Zhang, Y. Li, Z. Zhou, J. Yang, and F. Qi. 2017. Experimental and kinetic modeling study of laminar coflow diffusion methane flames doped with iso-butanol. Proc. Combust. Inst. 36 (1):1259. doi:https://doi.org/10.1016/j.proci.2016.06.111.
- Kizaki, Y., H. Nakamura, T. Tezuka, S. Hasegawa, and K. Maruta. 2015. Effect of radical quenching on CH4/air flames in a micro flow reactor with a controlled temperature profile. Proc. Combust. Inst. 35 (3):3389. doi:https://doi.org/10.1016/j.proci.2014.07.030.
- Knyazev, V. D., and I. R. Slagle. 1996. Experimental and theoretical study of the C2H3-H+C2H2 reaction. Tunneling and the shape of Falloff Curves. J. Phys. Chem. 100:16899. doi:https://doi.org/10.1021/jp953218u.
- Köhler, M., P. Oßwald, H. Xu, T. Kathrotia, C. Hasse, and U. Riedel. 2016. Speciation data for fuel-rich methane oxy-combustion and reforming under prototypical partial oxidation conditionsChem. Eng. Sci. 139:249. doi:https://doi.org/10.1016/j.ces.2015.09.033.
- Li, Q., T. Wang, Y. Liu, and D. Wang. 2012. Experimental study and kinetics modeling of partial oxidation reactions in heavily sooting laminar premixed methane flames. Chem. Eng. J. 207–208:235. doi:https://doi.org/10.1016/j.cej.2012.06.093.
- Liu, P., H. Lin, Y. Yang, C. Shao, B. Guan, and Z. Huang. 2015. Investigating the role of CH2 radicals in the HACA mechanism. J. Phys. Chem. A 119:3261. doi:https://doi.org/10.1021/jp5124162.
- Mai, T. V. T., M. V. Duong, X. T. Le, L. K. Huynh, and A. Ratkiewicz. 2014. Direct ab initio dynamics calculations of thermal rate constants for the CH4+O2=CH3+HO2 reaction. Struct. Chem. 25 (5):1495. doi:https://doi.org/10.1007/s11224-014-0426-2.
- Marinov, N. M., W. J. Pitz, C. K. Westbrook, M. J. Castaldi, and S. M. Senkan. 1996. Modeling of aromatic and polycyclic aromatic hydrocarbon formation in premixed methane and ethane flames. Combust. Sci. Technol. 116–117 (1–6):211. doi:https://doi.org/10.1080/00102209608935550.
- Maruta, K., T. Kataoka, K. N. Il, S. Minaev, and R. Fursenko. 2005. Characteristics of combustion in a narrow channel with a temperature gradient. Proc. Combust. Inst. 30 (2):2429. doi:https://doi.org/10.1016/j.proci.2004.08.245.
- Melton, T. R., A. M. Vincitore, and S. M. Senkan. 1998. The effects of equivalence ratio on the formation of polycyclic aromatic hydrocarbons and soot in premixed methane flames. Proc. Combust. Inst. 27 (2):1631. doi:https://doi.org/10.1016/S0082-0784(98)80001-5.
- Metcalfe, W. K., S. M. Burke, S. S. Ahmed, and H. J. Curran. 2013. A hierarchical and comparative kinetic modeling study of C1-C2 hydrocarbon and oxygenated fuels. Int. J. Chem. Kinet. 45 (10):638. doi:https://doi.org/10.1002/kin.20802.
- Milcarek, R. J., H. Nakamura, T. Tezuka, K. Maruta, and J. Ahn. 2019. Microcombustion for micro-tubular flame-assisted fuel cell power and heat cogeneration. J. Power Sources 413:191. doi:https://doi.org/10.1016/j.jpowsour.2018.12.043.
- Miller, J. A., and S. J. Klippenstein. 2004. The H+C2H2(+M)⇄C2H3(+M) and H+C2H2(+M)⇄C2H5(+M) reactions: Electronic structure, variational transition-state theory, and solutions to a two-dimensional master equationy. Phys. Chem. Chem. Phys. 6:1192. doi:https://doi.org/10.1039/B313645K.
- Miller, J. A., and C. F. Melius. 1992. Kinetic and thermodynamic issues in the formation of aromatic compounds in flames of aliphatic fuels. Combust. Flame 91 (1):21. doi:https://doi.org/10.1016/0010-2180(92)90124-8.
- Moskaleva, L. V., A. M. Mebel, and M. C. Lin. 1996. The CH3+C5H5 reaction: A potential source of beneze at high temperatures. Proc. Combust. Inst. 26 (1):521. doi:https://doi.org/10.1016/S0082-0784(96)80255-4.
- Nakamura, H., S. Suzuki, T. Tezuka, S. Hasegawa, and K. Maruta. 2015. Sooting limits and PAH formation of n-hexadecane and 2,2,4,4,6,8,8-heptamethylnonane in a micro flow reactor with a controlled temperature profile. Proc. Combust. Inst. 35 (3):3397. doi:https://doi.org/10.1016/j.proci.2014.05.148.
- Nakamura, H., R. Tanimoto, T. Tezuka, S. Hasegawa, and K. Maruta. 2014. Soot formation characteristics and PAH formation process in a micro flow reactor with a controlled temperature profile. Combust. Flame 161 (2):582. doi:https://doi.org/10.1016/j.combustflame.2013.09.004.
- Nguyen, T. L., L. Vereecken, and J. Peeters. 2006. Quantum chemical and theoretical kinetics study of the O(3P)+C2H2 reaction: A multistate process. J. Phys. Chem. A 110:6696. doi:https://doi.org/10.1021/jp055961k.
- Onda, T., H. Nakamura, T. Tezuka, S. Hasegawa, and K. Maruta. 2019. Initial-stage reaction of methane examined by optical measurements of weak flames in a micro flow reactor with a controlled temperature profile. Combust. Flame 206:292. doi:https://doi.org/10.1016/j.combustflame.2019.04.044.
- Richter, H., and J. B. Howard. 2000. Formation of polycyclic aromatic hydrocarbons and their growth to soot—a review of chemical reaction pathways. Prog. Energy Combust. Sci. 26 (4–6):565. doi:https://doi.org/10.1016/S0360-1285(00)00009-5.
- Saiki, Y., and Y. Suzuki. 2013. Effect of wall surface reaction on a methane-air premixed flame in narrow channels with different wall materials. Proc. Combust. Inst. 34 (2):3395. doi:https://doi.org/10.1016/j.proci.2012.06.095.
- San Diego Mechanism web page, University of California at San Diego. 2016. Chemical-kinetic mechanisms for combustion applications. http://combustion.ucsd.edu
- Scire, J. J., R. A. Yetter, and F. L. Dryer. 2001. Flow reactor studies of methyl radical oxidation reactions in methane-perturbed moist carbon monoxide oxidation at high pressure with model sensitivity analysis. Int. J. Chem. Kinet. 33 (2):75. doi:https://doi.org/10.1002/1097-4601(200102)33:2<75::AID-KIN1000>3.0.CO;2-9.
- Selvaraj, P., P. G. Arias, B. J. Lee, H. G. Im, Y. Wang, Y. Gao, S. Park, S. M. Sarathy, T. Lu, and S. H. Chung. 2016. A computational study of ethylene–air sooting flames: Effects of large polycyclic aromatic hydrocarbons. Combust. Flame 163:427. doi:https://doi.org/10.1016/j.combustflame.2015.10.017.
- Sharma, S., and W. H. Green. 2009. Computed rate coefficients and product yields for c-C5H5 + CH3 ≤ products. J. Phys. Chem. A 113 (31):8871. doi:https://doi.org/10.1021/jp900679t.
- Shukla, B., and M. Koshi. 2012. A novel route for PAH growth in HACA based mechanisms. Combust. Flame 159 (12):3589. doi:https://doi.org/10.1016/j.combustflame.2012.08.007.
- Shukla, B., A. Miyoshi, and M. Koshi. 2010. Role of methyl radicals in the growth of PAHs. J. Am. Soc. Mass Spectrom. 21 (4):534. doi:https://doi.org/10.1016/j.jasms.2009.12.019.
- Skjøth-Rasmussen, M. S., P. Glarborg, M. Østberg, J. T. Johannessen, H. Livbjerg, A. D. Jensen, and T. S. Christensen. 2004. Formation of polycyclic aromatic hydrocarbons and soot in fuel-rich oxidation of methane in a laminar flow reactor. Combust. Flame 136 (1–2):91. doi:https://doi.org/10.1016/j.combustflame.2003.09.011.
- Slavinskaya, N. A., and P. Frank. 2009. A modelling study of aromatic soot precursors formation in laminar methane and ethene flames. Combust. Flame 156 (9):1705. doi:https://doi.org/10.1016/j.combustflame.2009.04.013.
- Smith, G. P., Y. Tao, and H. Wang 2016. Foundational fuel chemistry model version 1.0 (FFCM-1). http://nanoenergy.stanford.edu/ffcm1
- Takahashi, F., and I. Glassman. 1984. Sooting correlations for premixed flames. Combust. Sci. Technol. 37 (1–2):1. doi:https://doi.org/10.1080/00102208408923743.
- Tartakovsky, L., and M. Sheintuch. 2018. Fuel reforming in internal combustion engines. Prog. Energy Combust. Sci. 67:88. doi:https://doi.org/10.1016/j.pecs.2018.02.003.
- Wang, H. 2011. Formation of nascent soot and other condensed-phase materials in flames. Proc. Combust. Inst. 33 (1):41. doi:https://doi.org/10.1016/j.proci.2010.09.009.
- Xu, F., and G. M. Faeth. 2000. Structure of the soot growth region of laminar premixed methane/oxygen flames. Combust. Flame 121 (4):640. doi:https://doi.org/10.1016/S0010-2180(99)00170-4.
- Yang, X., X. Shen, J. Santer, H. Zhao, and Y. Ju 2017. HP-Mech. http://engine.princeton.edu/mechanism/HP-Mech.html
- Zhu, R., and M. C. Lin. 2001. The CH3 + HO2 reaction: First-principles prediction of its rate constant and product branching probabilities J. Phys. Chem. A 105 (25):6243. doi:https://doi.org/10.1021/jp010698i.