
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The use of energetic nanoparticles to tailor the properties of a base liquid fuel has attracted attention due to the possibility of decreasing fuel consumption and increasing control over the combustion process. In this study, the role of nanomaterials in the consumption of hydrocarbon fuel vapor is investigated using reactive molecular dynamics. Simulations are performed with aluminum and iron nanoparticles inside an n-heptane and oxygen gas mixture. The role of atomic charges on the dynamics of nanoparticle-hydrocarbon interactions is also investigated using different charge equilibration methods. Results show that both nanomaterials act as catalysts and enhance fuel decomposition. The decomposition of fuel molecules is initiated by dehydrogenation at the particle’s surface. This reaction path occurs significantly faster than the oxidation and pyrolysis paths observed for n-heptane in absence of nanoparticles. The oxidation in the presence of aluminum is characterized by more rapid particle heating and fragmentation compared to iron. Metal fragments further enhance the reactivity of the system due to a higher surface area available for reactions. The atomic charge distribution was found to affect the kinetics and reactivity of the system, showing that the non-bonded interactions influence the oxidation process. This study confirms that the use of nanomaterials is beneficial to accelerate the decomposition of fuel and that the combustion behavior of the selected hydrocarbon is strongly dependent on the type of nanomaterial used in combination with the base fuel.
Introduction
The urgent need to reduce pollutant emissions and improve the efficiency of fossil fuel combustion has increased the interest in the development of the so-called nanofuels. A nanofuel comprises a suspension of nano-sized particles, e.g., aluminum, boron, iron, and their compounds, in a base liquid fuel (Basu and Miglani Citation2016). The addition of nanomaterials allows for tailored properties of the resulting nanofuel with improved combustion characteristics compared to the base fuel, including increased volumetric energy density, enhanced catalytic activity, faster burning rates, lower ignition delay times and reduction of pollutant emissions (Basu and Miglani Citation2016; Khond and Kriplani Citation2016). Depending on the combination of nanomaterial and base fuel, single droplet combustion experiments (Basu and Miglani Citation2016; Pandey, Chattopadhyay, and Basu Citation2017) have shown a variety of phenomena driven by the presence of nanomaterials, ranging from bubble nucleation in the droplet, through nanoparticle agglomeration and shell formation, to droplet fragmentation. In some cases, e.g., Gan and Qiao (Citation2011), the single droplet combustion process showed well-defined stages associated with the evaporation of the liquid and burning of each of the components, including base hydrocarbon fuel, metal and surfactant combustion. In other cases, e.g., Javed, Baek, and Waheed (Citation2015), the process is more intricate and interactions between fragments of nanomaterial and fuel vapor may be expected. The latter is also the case when the nanofuel spray is completely pre-vaporized before reaching the reacting region, with the reacting mixture consisting of oxidizer, fuel vapor (including surfactants) and agglomerates of nanomaterials (Javed, Baek, and Waheed Citation2015; Zhu et al. Citation2021).
The combustion behavior of nanofuels is strongly affected by the choice of the nanomaterial-base fuel combination. Note that the choice of the nanomaterial also requires considerations on the toxicity of the agglomerates (Singh et al. Citation2010). Aluminum, is particularly appealing and widely used as both solid fuel and fuel additive for its high energy density, low cost and availability. The popularity of aluminum as a solid propellant stimulated significant efforts in understanding its oxidation and combustion behaviors, e.g., Levitas, Pantoya, and Watson (Citation2008), Overdeep et al. (Citation2019). Later research efforts were also focused on understanding the behavior of aluminum nanoparticles (AlNPs) in nanofuels (Javed, Baek, and Waheed Citation2015; Lucas et al. Citation2020). Experimental and numerical studies showed a range of phenomena during AlNP oxidation. At high heating rates, fast oxidation driven by the melt-dispersion mechanism has been observed, whereas at very low heating rates diffusion-controlled oxidation prevails (Levitas, Pantoya, and Watson Citation2008). Investigation of the oxidation mechanism of AlNP for particle diameters in the range 1 to 10 nm also identified the size of 4 nm to be a threshold under which particles vaporize by oxygen-etching, and above which particles are subject to heterogeneous oxidation (Overdeep et al. Citation2019). Nanofuels based on AlNP showed improved combustion performance for a range of base fuels (Allen et al. Citation2011; Basu and Miglani Citation2016; Sundaram, Yang, and Zarko Citation2015). Recent studies of hydrocarbon-Al nanofuel combustion demonstrated that the presence of AlNP could facilitate the hydrocarbon oxidation process through the release of highly reactive radicals, such as AlO and O radicals, which help initiate the fuel consumption process (Lucas et al. Citation2020).
In order to develop more understanding of the behavior of complex chemical systems, reactive Molecular Dynamics (MD) was developed as a trade-off between good accuracy and reasonable computational cost to study the behavior of large molecular systems (Senftle et al. Citation2016). ReaxFF force field was widely used in MD simulations to study AlNP oxidation (Hong and van Duin Citation2015), coating (Hong and van Duin Citation2016), catalytic cracking (Castro-Marcano and van Duin Citation2013), as well as oxidation of hydrocarbons (Chenoweth, van Duin, and Goddard Citation2008). Over the past decade, MD simulations extensively described nanoparticle oxidation mechanisms, especially with AlNP, supporting experimental observations on diffusion-driven oxidation (Chu et al. Citation2018), as well as melt and diffusion mechanisms (Wang et al. Citation2009). Charge-driven diffusion was also identified as a possible mechanism to facilitate the movement of atoms within the nanoparticle (Chakraborty and Zachariah Citation2014). Simulations of nanoparticles with size in the range 2–4 nm showed that pure AlNP melt occurs at lower temperatures than usual bulk aluminum (Liu, Wang, and Liu Citation2018), as a consequence of pre-melting phenomena at the particle surface. MD simulations of molten AlNP oxidation with varying ambient temperature and O2 concentration also showed the formation of chain-like structures and explosion phenomena (often called microexplosions) as additional mechanisms to enhance oxidation (Li et al. Citation2020). Transition from diffusion oxidation to fragmentation of the nanoparticle was observed with increasing relative velocity between the nanoparticle and the oxidizer (Chang, Chu, and Chen Citation2020). While single non-oxidized and passivated particle combustion is widely investigated, little work has been done on the interactions between nanoparticles and a base fuel on a molecular level. An investigation on the thermal decomposition of nitroglycerin in presence of aluminum showed that aluminum additives accelerate the decomposition of nitroglycerin due to the strong attraction between aluminum and oxygen (Zhao et al. Citation2021). It was also demonstrated that the decomposition and heat release rates are improved in the presence of aluminum. A catalytic contribution of aluminum particles to the thermal decomposition of hexanitrostilbene was also observed (Zhao et al. Citation2020). More recently, the ignition and combustion behavior of methane and non-oxidized AlNP was investigated (Wu et al. Citation2021). This study further supports the microexplosion process of bare AlNP, in addition to their catalytic effect on methane dissociation and combustion.
In this study, ReaxFF MD simulations of n-heptane combustion in the presence of nanoparticles are performed to gain further insight into the oxidation behavior of nanofuels, with a focus on the interaction between the base hydrocarbon fuel vapor and nanoparticles. Two different nanomaterials are investigated: AlNPs, which can be considered a reference material for energy applications, and iron nanoparticles (FeNPs), which are an emerging material for the development of low-carbon combustion (Bergthorson Citation2018). The objectives of the work are (i) to investigate the mechanisms of hydrocarbon-nanoparticle interactions, including the role of charge transfer in the diffusion of species within the nanomaterial, and (ii) to assess the role of the type of nanomaterial in the combustion dynamics of the nanoparticle-fuel vapor mixture. The combustion of FeNPs has not been extensively investigated yet, therefore this study also contributes to the development of a new understanding on the use of iron nanoparticles as energetic materials.
Methods
The interaction between nanoparticles and n-heptane combustion is investigated using reactive MD. Simulations are performed using the ReaxFF force field (van Duin et al. Citation2001), which allows for the formation and dissociation of bonds. The high accuracy of ReaxFF arises from the fact that the force field parameters are tuned using Quantum Mechanics (QM) computations. In a reactive MD simulation, the atomic charges are computed at each time step by minimizing the electrostatic energy of the system while satisfying the electroneutrality constraint (Sanderson Citation1951). The Charge Equilibration (QEq) method (Rappe and Goddard Citation1991) is one of the most widely used methods for the computation of atomic charges in MD. However, this method allows for long-range charge transfers to take place even between infinitely-separated atoms, thus leading to a nonphysical representation of charge transfers in non-conducting media (Chen and Martínez Citation2007). To compensate for the ideal conductor approximation introduced by the QEq method, other methods have been proposed such as the Split Charge Equilibration (SQE) (Mathieu Citation2007), Atom Condensed Kohn Sham density functional theory to the Second-order approximation (ACKS2) (Verstraelen et al. Citation2013) and the Charge Transfer with Polarization Current Equalization (QTPIE) (Chen and Martínez Citation2007) method. Such methods shield the charge transfers to closely interacting atoms. In this study, results obtained with the QEq (Rappe and Goddard Citation1991) and QTPIE (Chen and Martínez Citation2007) methods are compared with each other to investigate the impact of charge distribution on the interactions between fuel, oxidizer and metal particles.
MD simulations are performed in LAMMPS (Plimpton Citation1995) equipped with the USER-REAXC (Aktulga et al. Citation2012) package and QEq (Aktulga et al. Citation2012) and QTPIE (Kritikos and Giusti Citation2020) charge equilibration methods. Note that the QTPIE method has a formalism similar to QEq. Thus, the same ReaxFF force field (with no additional parameters) can be used with both charge equilibration methods with small differences in the computed atomic charges (Chen and Martínez Citation2007; Kritikos et al. Citation2022). The ReaxFF AlCHO-2016 (Hong and van Duin Citation2016) and ReaxFF FeCHO-2016 (Islam et al. Citation2015) force fields are used to simulate n-C7H16 and O2 interactions with the AlNP and FeNP, respectively. The equations of motion are integrated using the Velocity-Verlet algorithm and are progressed using a 0.1 fs timestep. Periodic boundary conditions are applied in all directions. To prepare the nanoparticles, large crystal structures are initially built. The aluminum crystal is constructed using an FCC unit cell with a 4.098 Å lattice parameter (Nakashima Citation2019). The iron crystal is based on a BCC unit cell with a 2.866 Å lattice parameter (Pepperhoff and Acet Citation2001). Afterwards, spherical nanoparticles are extracted from the crystals. Both AlNP and FeNP have a diameter of 3 nm (AlNP and FeNP consist of 791 atoms and 1243 atoms, respectively). Before setting the configuration of each system, the energies of the nanoparticle and gas mixture molecules are minimized with a conjugate gradient algorithm.
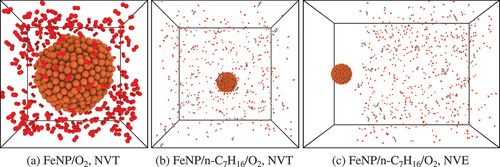
Simulations are performed with the canonical (NVT) and microcanonical (NVE) ensembles. NVT simulations are used to investigate pure nanoparticle oxidation and interactions between nanoparticle and n-C7H16/O2 gas mixture at a constant temperature. NVE simulations are used to gain more insight into the combustion transient with nanoadditives and gas mixture at different temperatures. summarizes the cases studied in this work. The NVT and NVE ensemble cases followed different preparation procedures. For the NVT simulations of nanoparticle oxidation without fuel, the nanoparticles and 300 O2 molecules are placed in cubic domains with side length equal to 50 Å. A screenshot of the domain for iron nanoparticles is shown in . The systems are then equilibrated at 300 K for 1 ns. For the fuel/oxygen gas mixture simulations, the nanoparticles are placed in cubic domains of 180 Å length surrounded by 30 n-C7H16 and 445 O2 molecules. shows a screenshot of the domain. The AlNP and FeNP systems are equilibrated at 500 K and 1000 K, respectively. Afterwards, they undergo a heating process to reach the target temperature of 1800 K. A constant heating rate of 10 K/ps is applied. During the simulations, the temperature of the system is held constant using the Nosé-Hoover thermostat with a damping constant of 100 fs. Results are obtained with both QEq and QTPIE charge equilibration methods.
Table 1. Simulation details.
The NVE simulations are performed with AlNPs and FeNPs interacting with 30 n-C7H16 and 445 O2 gas molecules. The nanoparticles and gas mixture are equilibrated independently for a total duration of 250 ps using the NVT canonical ensemble. The temperature of equilibration of each system is shown in . The gas mixture is equilibrated in a cubic domain with a length of 180 Å. After the equilibration procedure of the nanoparticles and gas mixture, the resulting systems are merged, leaving a 2 nm gap between the particle and the gas molecules in order to prevent any artificial interaction between them in this initial state. The initial configuration is shown in . The gas flow is observed to fill the 2 nm gap in less than 5 ps, which represents less than 0.5% of the total simulation time equal to 1 ns. Different temperature conditions for the gas and the nanoparticles were investigated in order to reproduce a cold solid nanoparticle entering a hot gas environment (see ). The procedure is similar to the study of Chang, Chu, and Chen (Citation2020). Note that prior to the equilibration stage, atomic velocities sampled from a Maxwell-Boltzmann distribution that matches the temperature of equilibration are assigned to all systems (both NVT and NVE ensembles). Also, during the equilibration and heating processes, C – O and H – O bonds are switched off to avoid any chemical reactions.
Figure 1. Initial domain configurations for (a)–(b) NVT simulations and (c) NVE simulations. CPK coloring is used to distinguish different chemical elements: Fe atoms are in orange; C atoms are in gray; O atoms are in red; H atoms are in white colour.
Results and discussion
NVT ensemble simulations
The impact of charge transfers on AlNP and FeNP oxidation dynamics is first investigated by comparing results obtained with QEq and QTPIE charge equilibration methods. Systems at a constant temperature of 300 K are considered. The number of O2 molecules and the evolution of the system’s potential energy is shown in . When the QEq method is used the absorption rate of O2 molecules significantly increases for both AlNP and FeNP nanoparticles. This is also indicated by the faster decrease in potential energy, which suggests an overall accelerated heat release. Note also that the O2 consumption in asymptotically reaches a higher limit when the QEq method is used compared to the results obtained with the QTPIE method. This suggests that the inward diffusion of oxygen atoms is hindered when the QTPIE method is applied. The role of charge transfers in the oxidation dynamics is further examined by comparing the atomic charge distribution predicted by the two charge equilibration methods. As shown in , the charges predicted by the QTPIE method are lower compared to the values found with the QEq method. This is a result of the shielding of the long-range charge transfers introduced by the QTPIE method. Henz, Hawa, and Zachariah (Citation2010) found that the oxidation of aluminum nanoparticles is primarily driven by an induced electric field between the positively charged metal core atoms and the negatively charged oxide shell. In the present study, simulations performed with the QEq method show larger charges for both the oxygen adsorbed on the surface and the nanoparticle metal core compared to results found with the QTPIE method. Such difference of charges affects the intensity of the induced electric field and therefore the diffusion and oxidation dynamics. Note also that the nanoparticles and O2 acquire a relatively high net charge when the QEq method is used due to the long-range charge transfers. This increases the electrostatic attraction between the negatively charged oxygen atoms and the positively charged metal atoms. As a consequence, the nanoparticle oxidation is further accelerated with the QEq method. Therefore, it can be concluded that the nonbonded interactions, i.e., van der Waals and Coulomb forces, contribute to a high degree in the oxidation and diffusion processes and should be carefully modeled to accurately predict nanofuel combustion.
Figure 2. Time evolution of consumed O2 (number of molecules) and Potential Energy (PE) during AlNP and FeNP oxidation without hydrocarbon fuel using QEq and QTPIE charge equilibration methods.
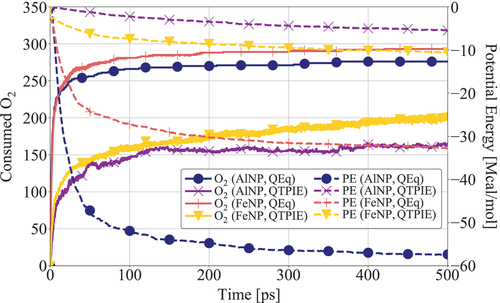
Figure 3. Reactant consumption in the presence and absence of nanoparticles using QEq and QTPIE methods. AlNP and FeNP simulations are conducted at K; gas-only simulations were performed at
K. The number fraction of each molecule corresponds to their number in each timeframe divided by the respective initial number.
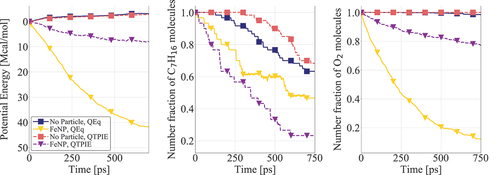
Table 2. Atomic charges of AlNP and FeNP computed with the QEq and QTPIE method at the beginning and end of the simulated time for a nanoparticle-oxygen system without hydrocarbon fuel. Charges are given in multiples of electron charge.
n-Heptane combustion simulations in the presence of FeNP are discussed next. It is noted that due to the high temperatures simulated here ( K) and the low melting point of aluminum (
K for 3 nm AlNP (Liu, Wang, and Liu Citation2018)) no AlNP high-temperature simulations were performed. The consumption of n-heptane and oxygen molecules for an ambient temperature of 1800 K is shown in . Note that gas-only simulations (without nanoparticles in the domain) at
K did not predict any reactions within the time period simulated in this work. Therefore, for the sake of analyzing the catalytic effect of FeNP, results of gas-only simulations at
K are reported. The addition of FeNPs in the gas mixture significantly increases the system’s reactivity. Specifically, at
K reactions occurred in the first 10 ps of the simulated time compared to the gas-only simulations where no reactions were observed. In addition, the nanoparticles increased the consumption rate of both fuel and oxidizer even compared to the gas-only simulations at
K. It can be concluded that FeNPs can act as catalysts and accelerate the kinetics of high-temperature combustion. The influence of the charge distribution on the absorption rate of oxygen is similar to the behavior observed for nanoparticle oxidation (). Specifically, the diffusion of oxygen molecules is hindered when charge transfers are limited to close interactions (QTPIE method) resulting in a decrease (about one-third lower) of absorbed O2 molecules at the end of the simulated time compared to results found when long-range charge transfers are enabled (QEq method). Nevertheless, the consumption of n-C7H16 increased with the QTPIE method. The reason for this behavior can be attributed to the fact that when the QEq method is used the higher absorption rate of O2 creates a thick oxide film on the particle surface that does not permit the fuel molecules to react with the metal atoms. In addition, the higher absorption of oxygen leads to fewer oxygen molecules available in the gas phase for the fuel to react with compared to the simulations where the QTPIE method is used, although the most dominant reaction pathway in the first stage of combustion is the one driven by the catalysis on the nanoparticle surface. The overall reactivity of the system is led by oxygen absorption and consumption, which is significantly faster in the simulation with the QEq method. This behavior is also demonstrated by the faster decrease in potential energy, suggesting higher reactivity in simulations performed with QEq method and FeNP (). It is evident that the choice of the charge equilibration method directly affects the overall kinetics and chemical evolution of the system. Note that in gas-only simulations at
K results show that the QTPIE method leads to a slightly slower consumption rate for both n-C7H16 and O2 molecules (). It can also be seen that n-C7H16 decomposition is initiated earlier using the QEq method compared to the QTPIE method. These differences become more evident in the presence of nanoparticles, making the prediction of charge transfers even more important when nanofuel combustion is studied.
Figure 4. Dissociation reaction of n-heptane molecules at a FeNP surface. The n-C7H16 dissociation is initiated by dehydrogenation. Colours indicate the atomic charges.
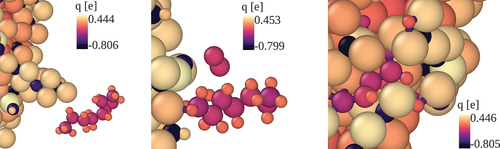
Simulations show that the hydrocarbon molecules undergo both dissociation and adsorption as they interact with the nanoparticle. The main interaction mechanism observed in the simulations consists in n-heptane molecules approaching the FeNP, driven by electrostatic attraction, followed by dehydrogenation and adsorption of fuel molecules on the FeNP surface. The overall adsorption process is shown in . Specifically, as the hydrocarbon molecules approach the FeNP, a Fe – H bond is formed. This subsequently leads to the dissociation of the H atom from the fuel molecule. Afterwards, the H atom is absorbed by the FeNP. This C – H bond scission reaction is repeated for the other H atoms, leading the C atoms to form bonds with neighboring Fe atoms. Finally, the carbon chain gets absorbed in the FeNP and later breaks down into smaller structures. This mechanism is responsible for the accelerated dynamics induced by the FeNP observed in Once the gas molecules get absorbed in the FeNP subsequent recombination with other species can take place leading to the formation of molecules on the nanoparticles’ surface. Formation of H2 and H2O molecules was observed at the FeNP surface. It is important to note that no major differences were found between the reaction pathways of n-heptane and FeNP predicted by the QEq and QTPIE charge equilibration methods.
NVE ensemble simulations
Figure 5. Temperature evolution for various systems comprising FeNP and AlNP.
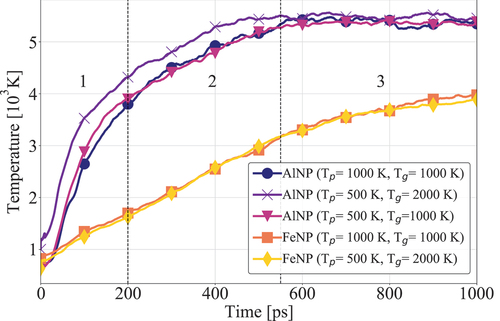
To give more insight into the transient combustion mechanism of a nanoparticle in a gas mixture of n-C7H16 and oxygen, simulations with the NVE canonical ensemble are discussed below. This section relies on results obtained with the QEq charge equilibration method only, since it offers overall accelerated kinetics (as shown in ), thus reducing the necessary computational time. The time evolution of the system’s temperature is shown in . The overall behavior of the system did not show significant variations for different initial temperature conditions of the gas mixture and nanoparticle. This suggests that the trajectory of the system is mainly dependent on the particle energy content and its release during the reaction dynamics. As observed in , the combustion process of AlNP systems can be divided into three stages. The first stage is characterized by a rapid heating. Subsequently, the heating rate decreases (second stage), followed by the convergence of the system’s temperature. During the first stage (i.e., below 0.2 ns of simulated time), AlNP combustion showcased a relatively high heating rate (). The heating rate appears to be in agreement with NVE simulations conducted by Liu, Wang, and Liu (Citation2018) for AlNP oxidation. This suggests that a very rapid particle ignition takes place. Following the ignition process, a large amount of heat is released, accounting for
80% of the temperature buildup (). The second stage (time period between 0.2 ns and 0.55 ns) is characterized by an approximately linear increase of the system’s temperature. In the third stage, the system reaches a steady-state combustion process at constant temperature
K.
Figure 6. Potential energy and n-heptane/oxygen consumption for systems containing AlNP and FeNP. Results are presented for cases with initial nanoparticle and gas temperatures equal to 1000 K. The number fraction of each molecule corresponds to their number in each timeframe divided by the respective initial number.
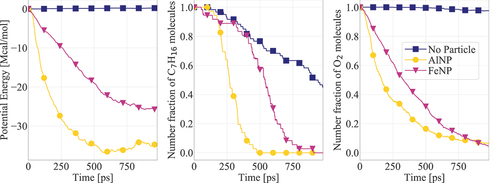
Analysis of the atomic trajectories shows that the first stage of significant temperature increase of AlNP systems () can be related to the bulk nanoparticle melt oxidation. The temperature rise observed in the first stage can be attributed to the exothermic nature of the oxidation process (Hong and van Duin Citation2015). The associated increase in heat release is also evident from the time evolution of potential energy shown in . The significant heat release melts the nanoparticle, a phenomenon that is in agreement with the studies of Li et al. (Citation2020) and Liu, Wang, and Liu (Citation2018). In most of the AlNP cases studied here, particle fragmentation was also observed. The particle breaks up into smaller clusters or forms metal chains. As a result, the reactivity of the system further increases due to the additional oxidation loci, i.e., increase of the particle’s reactive surface area. As the temperature continues to increase and after a small induction time, the chain growth is accompanied by microexplosions and related dispersion of particle clusters at velocities higher than 2 km.s−1. Such velocities are consistent with an explosion phenomenon (Li et al. Citation2020). The increased number of clusters allows for additional exposure of active material to the surrounding gas, further acting as an oxidation catalyst.
Systems containing FeNP demonstrate a less violent and more linear increase in the heat release rate before temperature stabilization (). No clear distinction between the first and second stages identified for AlNP systems can be made. This is because microexplosions, which typically characterize AlNP system dynamics, do not occur in the combustion of FeNP. In FeNP combustion, the initial stage of combustion (below 0.1 ns) corresponds to FeNP surface oxide film growth and surface pre-melt. This is followed by an approximately linear temperature increase until a constant value of K is reached. The lower energy density of iron might explain the less violent combustion.
Examination of the trajectories of the FeNP system shows that oxygen atoms approaching the nanoparticle surface at high relative velocity lead to local heating of the particle in the vicinity of the collision point. FeNP oxidation locally releases significant amounts of heat following Fe – O bond formation, generating hot spots that further facilitate the oxidation process. The phenomenon of microexplosion was not observed in the FeNP combustion. In fact, while chain growth followed by the fast fragmentation into clusters is observed in the case of AlNP, only mild evaporation of the FeNP is noticeable. This is likely to be a consequence of the iron’s higher melting point ( K for 3 nm FeNP (Fedorov, Shulgin, and Lavruk Citation2016)). The FeNP also conserves its initial structure throughout the early combustion stages.
While both AlNP and FeNP exhibit heterogeneous combustion behaviors, both nanoadditives drastically accelerate n-heptane and oxygen consumption. shows the time evolution of the fraction (with reference to the initial value) of the number of n-C7H16 and O2 molecules in the domain in the presence and absence of nanoadditives. While in absence of nanoparticles only reduced reactivity is observed during the first 1 ns of simulated time, the presence of AlNP or FeNP significantly accelerates the consumption of O2 and n-C7H16 molecules. Note that FeNPs seem to enable faster initiation of n-heptane dissociation. However, AlNPs lead to faster n-C7H16 and O2 consumption after reaction initiation. The faster reactant consumption in the cases of AlNP and FeNP further supports the catalytic role of the nanoparticles studied in this work.
Figure 7. Dominant intermediate species for the systems with AlNP (left) and FeNP (right). Results are presented for cases with initial nanoparticle and gas temperatures equal to 1000 K.
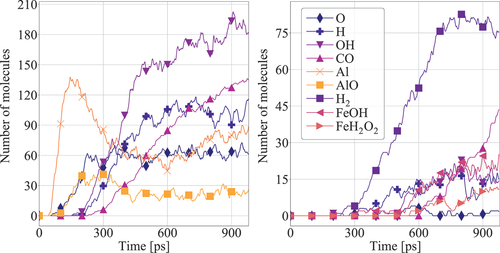
To further investigate the combustion mechanism and interaction of n-heptane with the nanoparticles, the time evolution of the number of molecules of selected reaction products is shown in . All species that, at any time step, exceed 10 molecules in the simulation box are reported. The combustion behavior with AlNP is analyzed first, (left). During the early combustion stages (i.e., in the first 0.2 ns of simulated time), free Al atoms are present in the gas phase in significant quantities. This observation is also an indicator of AlNP microexplosions taking place in the domain around 0.1 ns. The drastic decrease of free Al atoms after the first peak (, left) is related to Al oxidation and formation of clusters after individual Al atom oxidation. These results are consistent with the observation by Overdeep et al. (Citation2019) of particle melt vaporization. Smirnov et al. (Citation2015) recently conducted an experimental analysis of vaporized n-decane in the presence of 2.5% in mass of non-passivated AlNP. Their experiments revealed higher temperatures than pure n-decane combustion. Smirnov et al. (Citation2015) postulated that this effect could be related to the fast evaporation and release of pure Al clusters, free atomic oxygen and AlO radicals. These species were also advocated to enhance the chain reaction mechanism of n-decane. MD findings by Wu et al. (Citation2021) report that AlxOy clusters enhance hydrocarbon dissociation and combustion, further supporting the suggestions by Smirnov et al. (Citation2015). These results are in agreement with our analysis as the fast heat release and the number evolution of free oxygen and Al compounds are well correlated with n-heptane dissociation and consumption ().
The evolution of products when FeNPs are used is substantially different from combustion with AlNP. FeNP combustion products and intermediates only become significant after 200 ps of simulated time, as shown in (right). The addition of FeNP to n-heptane combustion produces a significant amount of H2 molecules compared to the case of AlNP. The generation of H2 molecules could be linked to the low atomic oxygen concentration in the simulation box (see ). On the contrary, in the AlNP case, a higher concentration of atomic oxygen is observed, which could favor the formation of OH. It should also be noted that in the simulations with FeNP, only traces of atomic Fe were found, whereas in the simulations with AlNP the concentration of atomic Al is more significant. This indicates that the evaporation process of FeNP is significantly slower compared to AlNP, which gives further evidence of the absence of microexplosion phenomena in FeNP systems.
Conclusions
The effect of aluminum and iron nanoparticles on the combustion of n-heptane and oxygen gas mixtures has been investigated using reactive molecular dynamics simulations. Two different charge equilibration methods, namely QEq and QTPIE methods, have been used to investigate the role of charge transfers on the combustion behavior.
Results show that charge transfers have an important role in the reactivity of the systems since simulations with different charge equilibration methods predict different reaction dynamics. The higher atomic charges predicted by the QEq method, compared to the charges found with the QTPIE method, lead to an overall acceleration of the reaction kinetics for all investigated systems. Both Al and Fe nanoparticles showed a catalytic effect on n-heptane decomposition, also accelerating oxygen consumption through oxidation of both the nanomaterial and products of the decomposed fuel. However, the reaction dynamics is strongly dependent on the selected nanomaterial. AlNP revealed a three-stage combustion phenomenon with microexplosions that increase the reactivity of the system through interactions of the ejected Al clusters with n-heptane. FeNP demonstrated a lower heat release with no clear identification of the microexplosion accelerated oxidation. It was observed that n-heptane dissociation is initiated by dehydrogenation when the fuel molecule gets in contact with the nanoparticle. Results of the present work suggest that the combination of hydrocarbons with selected nanomaterials could lead to significantly different combustion dynamics compared to the oxidation of the base fuel, opening up new possibilities for the design of novel fuels.
Authors’ contributions
Conceptualization: EMK, AG; Methodology: MSA, EMK; Formal analysis and investigation: MSA; Writing – original draft preparation: MSA; Writing – review and editing: EMK, AG; Supervision: EMK, AG.
Acknowledgements
Part of the research leading to these results was conducted while MSA was attending the Advanced Mechanical Engineering (AME) MSc programme at Imperial College London, Department of Mechanical Engineering. The support from the AME programme is acknowledged.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Aktulga, H. M., J. C. Fogarty, S. A. Pandit, and A. Y. Grama. 2012. Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques. Parallel. Comput. 38 (4– 5):245–59. doi:10.1016/j.parco.2011.08.005.
- Allen, C., G. Mittal, C.-J. Sung, E. Toulson, and T. Lee. 2011. An aerosol rapid compression machine for studying energetic-nanoparticle-enhanced combustion of liquid fuels. Proc. Combust. Inst. 33 (2):3367–74. doi:10.1016/j.proci.2010.06.007.
- Basu, S., and A. Miglani. 2016. Combustion and heat transfer characteristics of nanofluid fuel droplets: A short review. Int. J. Heat Mass Tran. 96:482–503. doi:10.1016/j.ijheatmasstransfer.2016.01.053.
- Bergthorson, J. M. 2018. Recyclable metal fuels for clean and compact zero-carbon power. Prog. Energ. Combust. 68:169–96. doi:10.1016/j.pecs.2018.05.001.
- Castro-Marcano, F., and A. C. T. van Duin. 2013. Comparison of thermal and catalytic cracking of 1-heptene from reaxff reactive molecular dynamics simulations. Combust. Flame 160 (4):766–75. doi:10.1016/j.combustflame.2012.12.007.
- Chakraborty, P., and M. R. Zachariah. 2014. Do nanoenergetic particles remain nano-sized during combustion? Combust. Flame 161 (5):1408–16. doi:10.1016/j.combustflame.2013.10.017.
- Chang, X., Q. Chu, and D. Chen. 2020. Shock-induced anisotropic metal combustion. J. Phys. Chem. C 124 (24):13206–14. doi:10.1021/acs.jpcc.0c02876.
- Chen, J., and T. J. Martínez. 2007. Qtpie: Charge transfer with polarization current equalization. A fluctuating charge model with correct asymptotics. Chem. Phys. Lett. 438 (4):315–20. doi:10.1016/j.cplett.2007.02.065.
- Chenoweth, K., A. C. T. van Duin, and W. A. Goddard. 2008. Reaxff reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J Phys. Chem. A 112 (5):1040–53. doi:10.1021/jp709896w.
- Chu, Q., B. Shi, L. Liao, K. H. Luo, N. Wang, and C. Huang. 2018. Ignition and oxidation of core-shell al/al2o3 nanoparticles in an oxygen atmosphere: Insights from molecular dynamics simulation. J. Phys. Chem. C 122 (51):29620–27. doi:10.1021/acs.jpcc.8b09858.
- Fedorov, A. V., A. V. Shulgin, and S. A. Lavruk. 2016. Study of iron nanoparticle melting. AIP Conf. Proc. 1770 (1):030099.
- Gan, Y., and L. Qiao. 2011. Combustion characteristics of fuel droplets with addition of nano and micron-sized aluminum particles. Combust. Flame 158 (2):354–68. doi:10.1016/j.combustflame.2010.09.005.
- Henz, B. J., T. Hawa, and M. R. Zachariah. 2010. On the role of built-in electric fields on the ignition of oxide coated nanoaluminum: Ion mobility versus Fickian diffusion. J. Appl. Phys. 107 (2):024901. doi:10.1063/1.3247579.
- Hong, S., and A. C. T. van Duin. 2015. Molecular dynamics simulations of the oxidation of aluminum nanoparticles using the reaxff reactive force field. J. Phys. Chem. C 119 (31):17876–86. doi:10.1021/acs.jpcc.5b04650.
- Hong, S., and A. C. T. van Duin. 2016. Atomistic-scale analysis of carbon coating and its effect on the oxidation of aluminum nanoparticles by reaxff-molecular dynamics simulations. J. Phys. Chem. C 120 (17):9464–74. doi:10.1021/acs.jpcc.6b00786.
- Islam, M. M., C. Zou, A. C. T. V. Duin, and S. Raman. 2015. Interactions of hydrogen with the iron and iron carbide interfaces: A reaxff molecular dynamics study. Phys. Chem. Chem. Phys 18 (2):761–71. doi:10.1039/C5CP06108C.
- Javed, I., S. W. Baek, and K. Waheed. 2015. Autoignition and combustion characteristics of heptane droplets with the addition of aluminium nanoparticles at elevated temperatures. Combust. Flame 162 (1):191–206. doi:10.1016/j.combustflame.2014.07.015.
- Khond, V. W., and V. M. Kriplani. 2016. Effect of nanofluid additives on performances and emissions of emulsified diesel and biodiesel fueled stationary ci engine: A comprehensive review. Renew. Sust. Energ. Rev. 59:1338–48. doi:10.1016/j.rser.2016.01.051.
- Kritikos, E., and A. Giusti. 2020. Reactive molecular dynamics investigation of toluene oxidation under electrostatic fields: Effect of the modeling of local charge distribution. J. Phys. Chem. A 124 (51):10705–16. doi:10.1021/acs.jpca.0c08040.
- Kritikos, E., A. Lele, A. C. van Duin, and A. Giusti. 2022. A reactive molecular dynamics study of the effects of an electric field on n-dodecane combustion. Combust. Flame 244:112238. doi:10.1016/j.combustflame.2022.112238.
- Levitas, V. I., M. L. Pantoya, and K. W. Watson. 2008. Melt-dispersion mechanism for fast reaction of aluminum particles: Extension for micron scale particles and fluorination. Appl. Phys. Lett. 92 (20). doi:10.1063/1.2936855.
- Li, G., L. Niu, W. Hao, Y. Liu, and C. Zhang. 2020. Atomistic insight into the microexplosion-accelerated oxidation process of molten aluminum nanoparticles. Combust. Flame 214:238–50. doi:10.1016/j.combustflame.2019.12.027.
- Liu, J., M. Wang, and P. Liu. 2018. Molecular dynamical simulations of melting al nanoparticles using a reaxff reactive force field. Mater. Res. Express 5 (6):065011. doi:10.1088/2053-1591/aac653.
- Lucas, M., S. J. Brotton, A. Min, C. Woodruff, M. L. Pantoya, and R. I. Kaiser. 2020. Effects of size and prestressing of aluminum particles on the oxidation of levitated exo-tetrahydrodicyclopentadiene droplets. J. Phys. Chem. A 124 (8):1489–507. doi:10.1021/acs.jpca.9b10697.
- Mathieu, D. 2007. Split charge equilibration method with correct dissociation limits. J. Chem. Phys. 127 (22):224103. doi:10.1063/1.2803060.
- Nakashima, P. 2019. The crystallography of aluminum and its alloys. In The encyclopedia of aluminum and its alloys, Taylor & Francis: Boca Raton FL USA. doi:10.1201/9781351045636-140000245.
- Overdeep, K. R., C. J. Ridge, Y. Xin, T. N. Jensen, S. L. Anderson, and C. M. Lindsay. 2019. Oxidation of aluminum particles from 1 to 10 nm in diameter: The transition from clusters to nanoparticles. J. Phys. Chem. C 123 (38):23721–31. doi:10.1021/acs.jpcc.9b05564.
- Pandey, K., K. Chattopadhyay, and S. Basu. 2017. Combustion dynamics of low vapour pressure nanofuel droplets. Phys. Fluids 29 (7):074102. doi:10.1063/1.4991752.
- Pepperhoff, W., and M. Acet. 2001. The structure of iron, constitution and magnetism of iron and its alloys. Berlin Heidelberg, Berlin, Heidelberg: Springer Berlin Heidelberg. doi:10.1007/978-3-662-04345-5.
- Plimpton, S. 1995. Fast parallel algorithms for short-range molecular dynamics. J. Comp. Phys. 117(1):1–19.https://www.lammps.org
- Rappe, A. K., and W. A. Goddard. 1991. Charge equilibration for molecular dynamics simulations. J. Phys. Chem. 95 (8):3358–63. doi:10.1021/j100161a070.
- Sanderson, R. T. 1951. An interpretation of bond lengths and a classification of bonds. Science 114 (2973):670–72. doi:10.1126/science.114.2973.670.
- Senftle, T. P., S. Hong, M. M. Islam, S. B. Kylasa, Y. Zheng, Y. K. Shin, C. Junkermeier, R. Engel-Herbert, M. J. Janik, H. M. Aktulga, et al. 2016. The reaxff reactive force-field: Development, applications and future directions. npj Comput. Mater 2 (1):15011. doi:10.1038/npjcompumats.2015.11.
- Singh, N., G. J. Jenkins, R. Asadi, and S. H. Doak. 2010. Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano Rev. 1 (1):5358. doi:10.3402/nano.v1i0.5358.
- Smirnov, V. V., S. A. Kostritsa, V. D. Kobtsev, N. S. Titova, and A. M. Starik. 2015. Experimental study of combustion of composite fuel comprising n-decane and aluminum nanoparticles. Combust. Flame 162 (10):3554–61. doi:10.1016/j.combustflame.2015.06.011.
- Sundaram, D. S., V. Yang, and V. E. Zarko. 2015. Combustion of nano aluminum particles (review). Combust. Explos. Shock Waves 51 (2):173–96. doi:10.1134/S0010508215020045.
- van Duin, A. C. T., S. Dasgupta, F. Lorant, and W. A. Goddard. 2001. Reaxff: A reactive force field for hydrocarbons. J. Phys. Chem. A 105 (41):9396–409. doi:10.1021/jp004368u.
- Verstraelen, T., P. W. Ayers, V. Van Speybroeck, and M. Waroquier. 2013. ACKS2: Atomcondensed Kohn-Sham DFT approximated to second order. J. Chem. Phys. 138 (7). doi:10.1063/1.4791569.
- Wang, W., R. Clark, A. Nakano, R. K. Kalia, and P. Vashishta. 2009. Fast reaction mechanism of a core(Al)-shell (Al2O3) nanoparticle in oxygen. Appl. Phys. Lett. 95 (26):261901. doi:10.1063/1.3268436.
- Wu, B., F. Wu, P. Wang, A. He, and H. Wu. 2021. Ignition and combustion of hydrocarbon fuels enhanced by aluminum nanoparticle additives: Insights from reactive molecular dynamics simulations. J. Phys. Chem. C 125 (21):11359–68. doi:10.1021/acs.jpcc.1c01435.
- Zhao, Y., Z. Mei, F.-Q. Zhao, S.-Y. Xu, and X.-H. Ju. 2021. Insight into the combustion mechanism of nitroglycerin/nano-aluminum composite materials. Struct. Chem. 32 (1):387. doi:10.1007/s11224-020-01640-7.
- Zhao, Y., J.-S. Zhao, F.-Q. Zhao, S.-Y. Xu, and X.-H. Ju. 2020. Revealing the decomposition behavior of hexanitrostilbene and aluminum nanoparticles composites: A reactive molecular dynamics simulation. Acta Astronaut. 177:320–31. doi:10.1016/j.actaastro.2020.07.042.
- Zhu, B., W. Chen, Y. Sun, B. Dai, and J. Liu. 2021. Ignition and combustion characteristics of al/n-heptane nanoslurry fuel droplets via a laser-ignition model. J. Energy Eng. 147 (6):04021057. doi:10.1061/(ASCE)EY.1943-7897.0000804.