Abstract
Ruthenium is a major fission product element among the platinum group elements (PGEs) in high-level liquid waste (HLLW). Ru tetra-oxide, RuO4, has high vapor pressure, which is high enough to be run off from its solution even at room temperature. Electrochemical oxidation method, to oxidize nitrosyl ruthenium to the tetra-oxide and then to remove ruthenium from liquid phase to gas phase, was studied to separate Ru from the HLLW. The advantage of this method requires neither additional reagents nor adjustment its valency before the oxidation and disadvantage is necessity of long time for oxidation. In order to improve oxidation rate, we carried out the experiments to clarify the effects following fundamental conditions to the electrochemical oxidation, which are (a) electrolyte temperature, (b) presence of promoter elements, (c) evaporation or reflux of condensed phase, and (d) using or not using of diaphragm at counter electrode. We found the fast oxidation conditions as follows: (1) higher temperature; 95°C, (2) Ce coexistence; 3000 ppm; and (3) usage of a diaphragm for counter electrode. However, evaporation or reflux conditions did not directly affect the electrochemical oxidation efficiency.
1. Introduction
A vitrification process for geological disposal of high-level liquid waste (HLLW) is one of the key technologies to accomplish the nuclear fuel cycle. Many elements (most of them are fission products) in HLLW should be dissolved in glass. Platinum group metals, Ru, Rh, and Pd, could precipitate as alloy and/or oxide particles in melting glass, and settled in the bottom of the glass melter, where platinum group metals are concentrated, making glass with high platinum-group metal content.
Ru especially, makes a needle-shaped oxide, which is a rutile type crystal, and forms a three-dimensional network at the bottom of the melter. Such glass becomes highly viscose and does not fall readily from the melter [Citation1], disturbing stable operation of the vitrification process. If the Ru concentration in HLLW could be lowered, then the vitrification process operation would become easier and more stable. We studied a method to remove Ru from the HLLW before vitrification.
There are many techniques reported to remove Ru from the HLLW, such as electro-deposition [Citation2,Citation3], volatilization [Citation4] in boiling nitric acid, and adsorption on a polymer gel [Citation5]. These techniques, however, miss a large portion of Ru or have long preparation times in order to unify ionic species before separation. Motojima [Citation6] reported using the electrochemical method to oxidize Ru as tetra-oxide, whose partial pressure is high enough to escape the solution at room temperature. This method is easy and quickly removes Ru from the solution. He applied constant current electrolysis at 1 A and 2.3–3.5 V. Such high potentials corroded the platinum electrode. To apply this technique to an actual separation process requires large surface-area electrodes. The purpose of this study is to improve efficiency and oxidation rate electrochemically under a lower potential. If the oxidation rate and/or efficiency for this method are improved, the surface area of the oxidation electrodes can be reduced.
The electrical potential for oxidation of Ru was first decided by measurements from a cyclic voltammogram (CV) and basic data for designing an electric cell, as well as the HLLW electrolyte for electrochemical oxidation of Ru were obtained.
2. Experimental
2.1. Electrochemical oxidation potential
A solution of nitrosyl ruthenium (RuNO3+) nitrate (Ru concentration 2.5 × 103 mg/L = 25 mM) in the nitric acid medium (3 M) was obtained from Furuya Metal Co. Ltd. The electrolyte cell was set on a heater controlled by a thermocouple set in the cell which measures the temperature of the solution. The working electrode was a platinum plate (10 mm × 20 mm) and the counter electrode was platinum wire (1 mmφ) in a glass tube with glass frit as diaphragm to avoid direct contact with the inside of the glass tube and the outside electrolyte solution in the glass tube. A Salt bridge, Luggin capillary, filled with the same Ru solution in the cell, was used and connected to the beaker where a reference electrode, Ag/AgCl, was set in the same Ru solution. The CV was measured with an HZ-3000, Hokuto Denko Corp., by changing the temperature of the solution in the cell from room temperature (50, 65, 80, and 95°C).
2.2. Electrochemical oxidation of ruthenium
shows the electrochemical oxidation cell's schematic. We prepared a round-bottomed flask with seven necked conical ground glass joints, for two electrodes, a condenser, thermocouple, Luggin capillary, sampling nozzle and air inlet (0.5 L/min). A nitrosyl ruthenium solution (150 mL, Ru: 3.0 × 103 mg/L in 3.0 M nitric acid) was heated to 50°C and 95°C with a heating mantle.
Figure 1. Schematic diagram of oxidation cell.
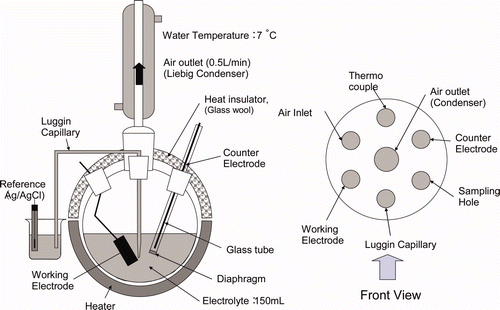
Working electrode and counter electrode were a 10 mm × 20 mm platinum plate and a 1 mmφ platinum wire, respectively. Electrochemical oxidation was carried out under the controlled potential electrolysis with 1.7 V (vs. SHE), using HZ-3000, Hokuto Denko Corp. A reference electrode was used Ag/AgCl with the saturated KCl solution as inner solution, which was set in a beaker connecting to the electrolyte cell with Luggin capillary. Electrolyte (2 mL) was taken at 1, 2, and 4 h after starting electrochemical oxidation. Samples for 6 h electrochemical oxidation were additionally taken if the difference of oxidation rate and efficiency were not be clarified. The ruthenium concentration was measured using an ICP-AES (ICPE-9000 Shimadzu Corp.).
Electrochemical oxidation conditions were changed with the following conditions, listed in
| 1. | Electrolyte temperature (Compared experimental results between condition A and B in Table 1). Electrolyte temperature was controlled to 50°C and 95°C, and the Ru removal rate for both temperatures was compared. | ||||
| 2. | Presence of a promoter element, cerium (Ce) (Compared experimental results between condition A and C). Ce(NO3)3·6H2O (Wako Pure Chemical Industries, Ltd.) was dissolved in the Ru solution, to adjust its concentration to 3.0 × 103 mg/L. | ||||
| 3. | Reflux or evaporation at 95°C (Compared experimental results between condition A and D). In evaporation mode, electrolyte volume and acidity was adjusted to be the same as the reflux conditions by adding 0.01 M of fresh nitric acid every 1 h as the same volume of solution evaporated during the experiment. The volume of vaporized liquid was measured by the volume of condensed water collected in a measuring cylinder, connected after the condenser ().
Figure 2. Schematic diagram of oxidation cell and condensate recovery for evaporation test. 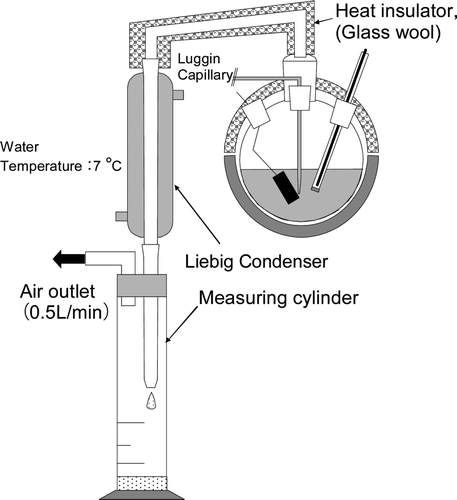 | ||||
| 4. | Influence of a diaphragm (Compared experimental results between condition A and E). Counter electrode, CE, was changed to a 10 mm × 20 mm platinum plate the same shape as the working electrode, WE, without a sintered glass diaphragm. | ||||
3. Results and discussion
3.1. Electrochemical oxidation potential
Ruthenium oxidation/reduction potentials between several Ru ion species were calculated based on thermodynamics of ruthenium data reviewed by Rard [Citation7] are listed in . Potentials of oxidation from lower valent Ru to hexavalent, heptavalent and octavalent are from 1.6–2.0, 1.3–1.6 and 1.2–1.4 V (vs. SHE), respectively. If the oxidation potential is selected higher than 1.6 V (vs. SHE), Ru is also oxidized via hexavalent and/or heptavalent to octavalent, RuO4. Therefore, higher oxidation potential is advantageous to achieve faster oxidation.
Table 1. Experimental conditions of electrochemical oxidation.
Table 2. Relative oxidation/reduction potential for ruthenium species (vs. SHE).
Table 3. Oxidation/reduction potentials(V) for Ru valency.
shows CV of ruthenium in nitric acid solution and 3 mol/L of nitric acid as blank solution at 25°C. The cathode current is increasing from the oxidation potential is the 1.1 V(vs Ag/AgCl), which is the oxidation to RuO4. The cathode current peak, Epc, height at 1.50 V (vs Ag/AgCl) is much larger than that of anode current peak, Epa, height at 1.32 V. The reaction occurred at 1.50 V should be an irreversible reaction. Ru ion in the solution is oxidized to octavalent as RuO4. The vapor pressure of RuO4 is high enough to escape from a solution, into gaseous phase even at room temperature. This suggests that the reaction at 1.50 V (vs. Ag/AgCl) should be irreversible and should be produced RuO4. There are also anode and cathode peaks at 0.98V and 0.91V in Figure 3, respectively. Compared with potential data in Table 2, oxidation-reduction reactions of ruthenium species are listed in Table 3.
Figure 3. Cyclic voltammogram at 25°C.
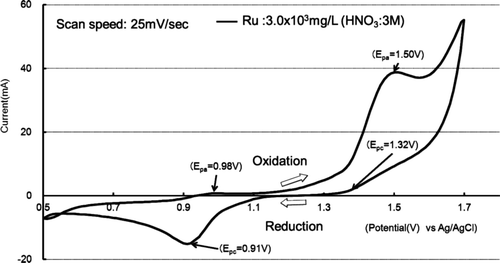
Taking fast oxidation rate into consideration, we selected 1.5 V (vs. Ag/AgCl) as an oxidation potential for Ru oxidation and removal from the solution.
We also measured CV of ruthenium solution at 25, 50, 70, and 95°C as shown in . The cathode peak at 1.5 V and 25°C was shifted to a lower potential as temperature increases. The higher temperature seems to be achieving faster oxidation even at the same potential.
Figure 4. Cyclic voltammogram at 25, 50, 70 and 95°C.
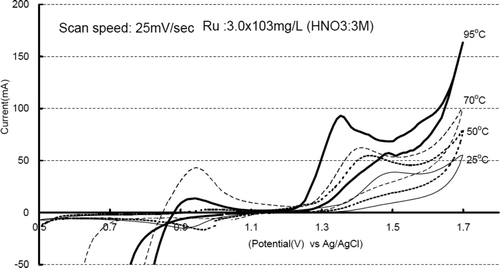
3.2. Electrochemical oxidation
Ruthenium ions might be simply oxidized electrochemically on the electrode. This means that the reaction rate depends on its concentration, as a first-order reaction. Therefore, higher Ru concentration has advantage in removal rate. The removal rate can be written as follows;
shows the relation of Ru concentration and time of electrolysis for all conditions. Ninety-five percent confidence interval was obtained for 6% of Ru concentration with duplicate measurement, which is also indicated in the figure. Each plot indicates the Ru concentration at each sampling time. Rate constant, k 1 and relation coefficient, r 2 for each condition were determined with least square method and listed in . The relation coefficient of the results at condition A and C, which are only Ru solution without any conditions, are bigger than 0.99. Therefore, the Ru oxidation process follows the Equation (3), it should be a first-order reaction.
Figure 5. Decreasing ratio of Ru concentration, [Ru] t /[Ru]0, with time.
![Figure 5. Decreasing ratio of Ru concentration, [Ru] t /[Ru]0, with time.](/cms/asset/4cedce36-8b48-423d-ba5f-45bf6d546c32/tnst_a_649084_o_f0005g.gif)
Table 4. Raction rate constant k 1 for each electrochemical oxidation condition.
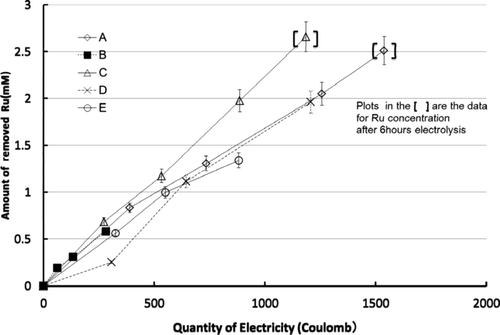
shows the increase of Ru removal amount from the solutions with the quantity of electricity for electrolytes at different conditions. The efficiency of the electrochemical oxidation process is defined as the molar amount of Ru removed for each coulomb, C.
Figure 6. Relation of amount of removed Ru and quantity of electricity.
3.2.1. Effect of electrolyte temperature: compared with condition between A and B
Based on , ruthenium removal rate at 95°C (A) is faster than that at 50°C (B) compared with its concentration at 6 h of electrochemical oxidation. Rate constant at 95°C is four times larger than at 50°C ().
Since the slopes of the two lines in the Figure 6 are nearly the same, the efficiency, however, has not been changed even for different temperatures.
3.2.2. Presence of promoter element, cerium: compared with condition between A and C
Cerium is a major fission product, whose concentration in high active waste solution is nearly the same as Ru [Citation8]. Cerium(III) nitrate was added to the Ru solution at a concentration of 3.0 × 103 Ce mg/L. Ce(III) is also oxidized during electrochemical oxidation to a tetravalent state. It is known that Ce promotes Ru oxidation with electrochemical oxidation process at nearly room temperature [Citation6].
Chemical reactions for RuNO3+ and Ce and the oxidation rate for RuNO3+ are written as follows,
shows the decreasing ruthenium concentration with time at 95°C without Ce (A) and with Ce (C). Comparing with oxidation rate of both conditions, there are no significant differences between them. Motojima [Citation6] reported that Ce works as a promoter, which improves the oxidation rate of Ru. We confirmed that this cannot be applicable at higher temperature. This should be explained that direct Ru oxidation reaction, Equation (4), rate was increased with temperature more than that Ce oxidation, Equation (5).
Concerning of oxidizing efficiency, i.e. the slope of the lines of , Ce is effective to increase oxidization rate of Ru. The difference in values for efficiency is stemmed from the second term in the right-hand side of Equation (9): k3 [RuNO3+][Ce4+] n .
This term should be more effective with a lower RuNO3+ concentration. Because Ce4+ concentration increases, low oxidation Ru species decrease. Therefore, the second term of k3 [RuNO3+][Ce4+] n becomes relatively larger than the first term of k1 [RuNO3+]. Therefore, the existence of Ce is advantageous from oxidation efficiency.
3.2.3. Evaporation: compared with condition between A and D
Treatment of vaporized liquid must be considered in the design of the electrolyte cell for the actual process operation under high temperature, such as 95°C. Some of the vaporized liquid was returned as condensate with dissolved gaseous RuO4. Therefore, the influence of returning condensate was estimated. The experimental diagram is shown in . Evaporated gas was cooled and condensed at the condenser and liquid was flown into the measurement cylinder to measure the volume of evaporated liquid. The same volume of fresh nitric acid as evaporated liquid measured by the cylinder (0.01 M) was added to keep the same electrolyte volume and acidity in the experiment in evaporation mode.
Compared with line A and D in the Figure 5, these lines are nearly the same as that for reflux and evaporation. Therefore, evaporation does not influence the removal of Ru. Actually, the total condensate volume was approximately 80 mL in 6 h for this experimental system and its Ru concentration is less than 150 mg/L, where Ru concentration was 0.12 mM in condensate at its highest. Compared with the 2 mM which was removed from the electrolyte of this experiment, the Ru amount absorbed in condensate was small. Furthermore, Ru in the condensate was RuO4(aq), that did not influence electro oxidation. Contact time between the liquid and RuO4 was only 1.8 s, since the inside of the condenser volume was 16 mL (20 cm × φ1.0 cm) and flow rate was 0.5 L/min.
Considering the actual plant, the contact time should be much longer than that obtained in this experiment. If dissolution of RuO4 into nitric acid follows Henry's law [Citation9], the Ru concentration in the condensate solution can be calculated.
where
h: Henry's constant,
P(RuO4): Partial pressure of RuO4(g),
c(RuO4): Concentration of RuO4 in the solution.
The averaged RuO4 concentration was calculated from the amount of Ru removed (2 mM) and volume of purged air during electrochemical oxidation.
2 mM/(total gas volume) = 0.011 mM/L and partial pressure was 0.19 Torr, assuming RuO4 as an ideal gas.
Henry's constant is 100 at 0.1 M HNO3 and 40°C.
c(RuO4) = 0.19/100 = 1.9 mM.
The Ru amount is near the same as that removed by electrochemical oxidation. This does of course depend on the evaporation rate, oxidation efficiency and purging air volume.
Therefore, the treatment of evaporated gas for an actual plant should be carefully designed.
3.2.4. Using diaphragm for separating electrolytes: compared with condition between A and E
If a diaphragm is unnecessary, it would be easy to design an instrument for the actual process of an electrochemical cell and it would also be convenient to maintain the cell after operation.
Compared with line A and E in the , when the diaphragm was not used, the oxidation rate is obviously decreased. But efficiency without the diaphragm was nearly the same that as with the diaphragm, as shown in .
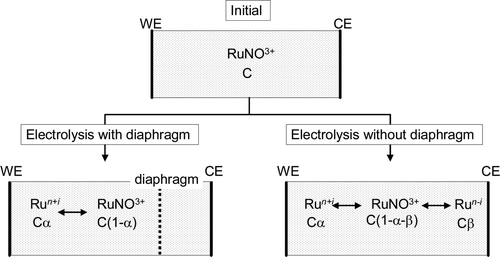
shows the ionic Ru species in the electrolyte during electrolysis with an initial ruthenium (RuNO3+) concentration of C mol and oxidation efficiency, α. With a diaphragm, RuNO3+ was just oxidized to higher oxidation states and the concentration of RuNO3+ was changed to C(1−α) and oxidized ruthenium species Ru n+i was Cα. But when the diaphragm was not used during electrolysis, Ru ionic species should be changed to oxidized Ru n+i and reduced Ru n−i , whose concentrations were Cα and Cβ. Therefore, the RuNO3+ concentration was to be C(1−α−β). Based on Equation (3), the rate of decrease of the RuNO3+ concentration is proportional to its concentration. Since C(1−α) is larger than C(1−α−β), using a diaphragm is advantageous to Ru removal.
Figure 7. Ru ionic species in electrolyte with and without diaphragm.
4. Conclusion
A fundamental study for an electrochemical oxidation method was carried out, in order to achieve efficient and fast ruthenium removal from a HLLW solution.
Oxidation potential was set at 1.5 V (vs. Ag/AgCl) = 1.7 V(SHE)] based on cyclic voltammogram of Ru nitric acid solution.
Electrochemical oxidation conditions for fast oxidation of ruthenium were achieved in following conditions: (1) higher temperature, (2) Ce coexistence, and (3) usage of a diaphragm for counter electrode. But evaporation or reflux conditions do not directly affect the electrochemical oxidation efficiency.
Acknowledgments
The authors thank Mr. M. Igarashi, Mr. H. Nemoto, Mr. S. Jitsukata and Mr. T. Kuno in the Process control analysis section at Tokai Reprocessing Center in JAEA for their accurate and precise analysis based on their careful preparation for measurement of ruthenium concentrations. The present study is the result of ‘Development of High-level Liquid Waste Conditioning Technology for Advanced Nuclear Fuel Cycle’ entrusted to ‘Japan Atomic Energy Agency’ by the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
Related Research Data
References
- Sundaram , S.K. and Perez , J.M. Jr. September 2000 . Nobel metals and spinel settling in high level waste glass melters Letter Report PNNL-13347
- Ozawa , M. , Suzuki , T. , Koyama , S. and Fujii , Y. 2005 . Separation of rare metal fission products in radioactive wastes in new directions of their utilization . Prog. Nucl. Energy , 47 : 462 – 472 .
- Jayakumar , M. , Venkatesan , K.A. and Srinivasan , T.G. 2005 . Feasibility studies on the electrochemical recovery of fission platinoids from high-level liquid waste . J. Radioanal. Nucl. Chem , 284 : 79 – 85 .
- Sato , T. 1989 . Volatilization behavior of ruthenium from boiling nitric acid . J. Radioanal. Nucl. Chem , 129 : 77 – 84 .
- Mimura , H. , Ohta , H. and Akiba , K. 2002 . Uptake and recovery of ruthenium by alginate gel polymers . J. Nucl. Sci. Technol , 39 : 655 – 660 .
- Motojima , K. 1990 . Removal of ruthenium from PUREX Process(II) . J. Nucl. Sci. Technol , 27 : 262 – 266 .
- Rard , J.A. 1985 . Chemistry and thermo dynamics of ruthenium and some of its inorganic compounds and aqueous species . Chem. Rev , 85 : 1 – 39 .
- Kuno , T. , Nakamura , Y. and Okano , M. Composition of insoluble materials in highly active liquid waste at Tokai Reprocessing Plant . Proceedings of GLOBAL2005 . Tsukuba , , Japan No. 208 (2005), 2005 [CD-ROM]
- Sasahara , A. and Kawamura , F. 1988 . Formation rate and gas–liquid equilibrium of RuO4 . J. Nucl. Sci. Technol , 25 : 472 – 478 .