Abstract
The chemical states of radioactive Cs (caused by Fukushima Daiichi Nuclear Power Plant accident) in the contaminated soils have been characterized by the desorption experiments using appropriate reagent solutions and size fractionation of the contaminated soils. More than 65% of radioactive Cs remained in the residual fraction of the soil samples after treatment of 1 mole L−1 NH4Cl solution and 1 mole L−1 CH3COOH solution. Approximately 70% of radioactive Cs in the residual fraction were associated with the size fractions larger than the elutriated one, even though mica-like minerals were present in the elutriated one. These results strongly suggest that radioactive Cs was irreversibly associated with soil components other than mica-like minerals in the contaminated soil.
1. Introduction
The nuclear accident at the Fukushima Daiichi Nuclear Power Plant (FDNPP) occurred as a consequence of the massive earthquake and associated tsunami that struck the Tohoku and north Kanto regions of Japan on 11 March 2011. A series of hydrogen explosions occurred from 13 March 2011 to 15 March 2011 at the units 1, 2, and 3. The release rate of 137Cs on 15 March 2011 is estimated between 1012 and 1015 Bq/h [1]. This fallout radioactive Cs was dispersed from FDNPP to ocean [1,2] and land [1]. Some of the released radioactive Cs was deposited on the ground of the area located in the northwest direction from FDNPP. The spatial concentration distribution and depth profiles of radioactive Cs were measured [3,4] to estimate the dose rate and to estimate the fate in the terrestrial environment.
For the estimation of migration of radioactive Cs, sorption behavior of the deposited radioactive Cs should be clarified. Cesium at low concentration is strongly sorbed on mica-like minerals [5–7]. These suggest that some amounts of radioactive Cs are irreversibly sorbed with soils. However, the characteristics of the irreversible sorption of fallout Cs in the contaminated soils are scarcely known.
In the present study, the desorption experiments of radioactive Cs using appropriate reagent solution have been carried out to clarify the association of radioactive Cs with soils. We also carried out the elutriation and sieve treatment for analyzing the size dependence of radioactive Cs sorption and the mineral composition. Based on the results, we discuss an appropriate remediation treatment of the contaminated soils.
2. Experimental
2.1. Materials and their radioactivity
Three different soil samples of soil A, soil B, and soil C were collected at Hiso, Iitate-mura, Fukushima prefecture, Japan on early April 2011. The soil samples were stored for about 4 months in a room at room temperature. The soil samples were dried in air for 5 days and were preliminary sieved to remove plant roots, litter, and gravels of more than about 2 mm. Radioactivities of 137Cs in the sieved soil and the solution samples were measured by γ-spectrometer (ORTEC) to estimate the desorption behaviors of radioactive Cs. The radioactive concentrations of 137Cs in the samples (Bq/kg) were determined based on the calibrated standard samples (DBA Isotope Products Laboratories, USA).
2.2. Desorption experiments
We carried out the desorption experiments in two different procedures. In the first procedure, the sieved soil samples of approximately 5 g merged with 1 mole L−1 NH4Cl solution of 40 mL for 24 h at 25°C. The NH4Cl solution was separated from the sieved soil samples by centrifugation for 10 min at 1700 rpm. The supernatant was filtered through membrane filter of 0.45 μm. The radioactivity of the solution was measured. This desorption process was repeated two times. The resulted NH4Cl-treated soil samples merged with 1 mole L−1 CH3COOH of pH ca. 2.4 of 40 mL. The CH3COOH solution was separated from the NH4Cl-treated soil samples by centrifuging for 10 min at 1700 rpm. The supernatant was filtered through membrane filter of 0.45 μm. The radioactivity of the solution was measured. The residual soil samples were dried in air, and the radioactivity was measured.
In the second procedure, the sieved soil samples of approximately 5 g merged with 1 × 10−3 mole L−1 H2SO4 solution (pH is ca. 2.7) of 40 mL for 24 h at 25°C. The H2SO4 solution was separated from the sieved soil samples by centrifugation for 10 min at 1700 rpm. The supernatant was filtered through membrane filter of 0.45 μm. The radioactivity of the solution was measured. This desorption process was duplicated. The treated soil samples merged in duplicate with 1 mole L−1 H2SO4 of 40 mL. The 1 mole L−1 H2SO4 solution was separated from the treated soil samples by centrifuging for 10 min at 1700 rpm. The supernatant was filtered through membrane filter of 0.45 μm. The radioactivity of the solution was measured. The residual soil samples were dried in air, and the radioactivity was measured. Note that the desorption experiments were carried out on August 2011, indicating that the contact time of radioactive Cs with soils was more than 5 months.
The same desorption experiments by the two different procedures were carried out at room temperature and at 60°C.
2.3. Elutriation and sieve treatment
The soil samples treated after 1 mole L−1 NH4Cl and the 1 mole L−1 CH3COOH solutions were elutriated with deionized water for 10–30 min. The supernatant solution was recovered and was elutriated in triplicate. The elutriated soil samples were dispersed in 1 mole L−1 NaCl solution and were separated from the solution by centrifuging for 30 min at 3000 rpm. The elutriated soil samples were recovered by using ethanol solution and dried at 40°C. Radioactivity of Cs in the elutriated soil samples was measured.
The precipitated soil by the elutriation was recovered and separated in different sizes of >300 μm, between 300 and 106 μm, and <106 μm by sieving. The radioactivity of sieved soil samples was measured using γ-spectrometer. The elutriated and sieved soil samples were analyzed by X-ray diffraction (XRD) for determining mineral compositions in the soils.
3. Results
3.1. Desorption of 137Cs from the soil samples by reagent solutions
Radioactive concentrations of the soil samples were 5.5 × 104 ± 5.2 × 103 Bq/kg, 1.1 × 105 ± 5.7 × 103 Bq/kg, and 8.7 × 104 ± 7.0 × 103 Bq/kg for soil A, soil B, and soil C, respectively. The desorbed fraction of 137Cs from the soil samples by 1 mole L−1 NH4Cl solution and 1 mole L−1 CH3COOH solution and that by 1 × 10−3 mole L−1 H2SO4 solution and 1 mole L−1 H2SO4 solution is shown in and , respectively. After the desorption experiment, the total amounts of 137Cs recovered in the desorbed and resided fractions were approximately 90% to the initial one, indicating that measurement error of the desorption experiments was approximately 10% in the present study.
Figure 1. Percent fractions of radioactive Cs desorbed from the soils A, B, and C by 1 mole L−1 NH4Cl solution and 1 mole L−1 CH3COOH solution, and residual one.
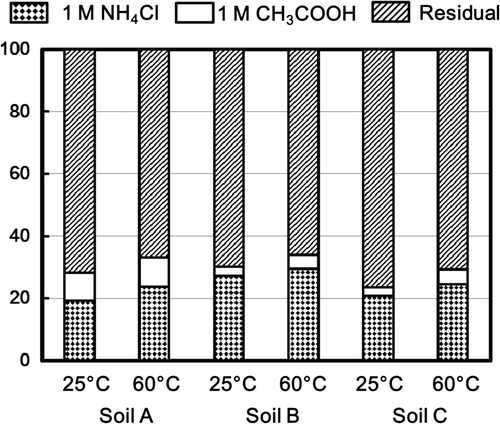
Figure 2. Percent fractions of radioactive Cs desorbed from the soils A, B, and C by 0.001 mole L−1 H2SO4 solution and 1 mole L−1 H2SO4 solution, and residual one. Arrows show fractions of Cs desorbed by 0.001 mole L−1 H2SO4 solution.
At 25°C, approximately 20%, 25%, and 20% of 137Cs were desorbed by 1 mole L−1 NH4Cl solution from soil A, soil B, and soil C, respectively. Between 2% and 8% of 137Cs were desorbed by 1 mole L−1 CH3COOH solution from three soil samples. More than 65% of 137Cs resided in the soil samples after treatment of 1 mole L−1 NH4Cl solution and 1 mole L−1 CH3COOH solution.
At 60°C, approximately 23%, 30%, and 25% of 137Cs were desorbed by 1 mole L−1 NH4Cl solution from soil A, soil B, and soil C, respectively. Between 4% and 7% of 137Cs were desorbed by 1 mole L−1 CH3COOH solution from three soil samples. Approximately 65% of 137Cs resided in the soil samples after treatment of 1 mole L−1 NH4Cl solution and 1 mole L−1 CH3COOH solution. These indicate that the desorption behavior of radioactive Cs at 60°C is nearly the same as that at room temperature.
shows that less than 1% of 137Cs was removed by 1 × 10−3 mole L−1 H2SO4 solution. Approximately 60%, 40%, and 39% of 137Cs were desorbed by 1 mole L−1 H2SO4 solution from soil A, soil B, and soil C, respectively. Approximately 40–60% of 137Cs resided in the soil samples after treatment with 1 × 10−3 mole L−1 H2SO4 solution and 1 mole L−1 H2SO4 solution.
3.2. 137Cs in different size fraction of soil components
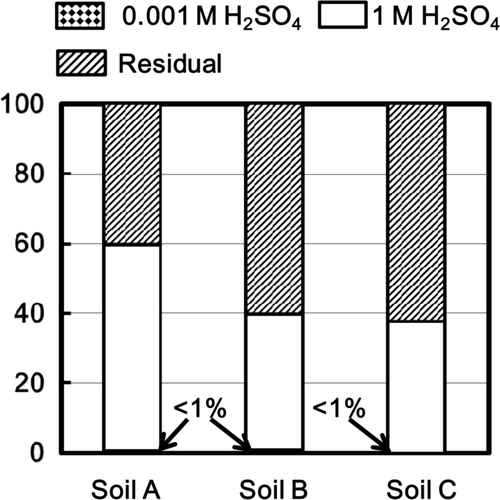
In the residual fractions of the soils after the treatment of 1 mole L−1 NH4Cl and 1 mole L−1 CH3COOH solutions, approximately 3%, 30%, and 14% of 137Cs were present in the elutriated fraction of soil A, soil B, and soil C, respectively (). Approximately 33%, 30%, and 50% of 137Cs were distributed in the fraction of the soil components larger than the elutriated one and less than 106 μm (hereinafter called as the fraction less than 106 μm) of soil A, soil B, and soil C, respectively. Approximately 35%, 18%, and 24% of 137Cs were distributed in the fraction between 106 and 300 μm of soil A, soil B, and soil C, respectively. About 30%, 23%, and 13% of 137Cs were present in the fraction larger than 300 μm of soil A, the B, and soil C, respectively. These results indicated that more than 95%, 70%, and 85% of 137Cs for soils A, B, and C, respectively, was distributed in the fractions other than the elutriated one.
Figure 3. Percent fractions of the resided radioactive Cs in the elutriated fraction, the settled fraction less than 106 μm, the fraction between 106 μm and 300 μm, and the fraction larger than 300 μm of the soils A, B, and C after the treatment of 1 mole L−1 NH4Cl solution and 1 mole L−1 CH3COOH solution.
In the elutriated fraction, the radioactive concentration of 137Cs was 4.3 × 106 Bq/kg, 5.8 × 106 Bq/kg, and 1.6 × 106 Bq/kg in soil A, soil B, and soil C, respectively. In soil A, the radioactive concentrations of 137Cs of the different size fractions higher than the elutriated fraction were 6.4 × 105 Bq/kg, 4.7 × 105 Bq/kg, and 2.1 × 105 Bq/kg in the fraction of the soil components larger than elutriated one and less than 106 μm, the fraction between 106 μm and 300 μm, and the fraction larger than 300 μm, respectively. Similar tendency was obtained in soil B and soil C. The radioactive concentration of 137Cs in the elutriated fraction was higher by approximately 10 times than the other fractions, indicating that 137Cs had high affinity to the elutriated fraction.
3.3. XRD patterns of different size fraction of soil components
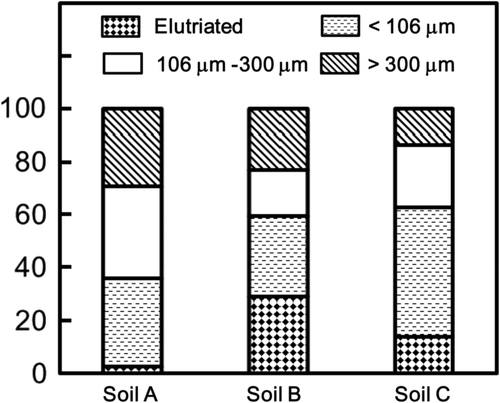
XRD patterns of the different size fractions () showed that distinct peaks observed higher angle than 20° in 2θ, indicating silicate minerals, aluminous minerals, feldspar, feldspathoid, sodalite, olivine, and borosilicate. These peaks were observed in all of the different size fractions in the three soils. Interestingly, the number of the distinguished peaks was the smallest in the elutriated fraction among the fractionated soils. These results are nearly the same among the three soils.
Figure 4. XRD patterns of the elutriated fraction (WE), the settled fraction less than 106 μm, the fraction between 106 μm and 300 μm, and the fraction larger than 300 μm of the soils A, B, and C after the treatment of 1 mole L−1 NH4Cl solution and 1 mole L−1 CH3COOH solution. Sm: smectite-like clay minerals, M: mica-like minerals, H: hornblende-like minerals, K: kaolinite-like minerals, Q: quartz, O: orthoclose, C: cristobalite, F: feldspar group, S: stishovite (SiO2), G: gibbsite-like minerals, SL: sodalite-like minerals, OL: olivine, SS: sorosilicate.
The peaks at angle less than 20° in 2θ were distinguished in the different size fractions. The peaks around 14°, 11°, 8°, and 6° in 2θ were assigned to kaolinite-like minerals (K), hornblende-like ones (H), mica-like ones (M), and smectite-like ones (Sm), respectively. The representative minerals and clay minerals in the fractions elutriated and sieved in the soils A, B, and C determined by XRD are summarized in .
Table 1. The representative minerals and clay minerals in the fractions elutriated and sieved in the soils A, B, and C determined by XRD.
In soil A, the peaks originated from kaolinite-like minerals were observed in all fractions. The peak of hornblende-like minerals was observed in the fractions less than 106 μm. The peak of mica-like minerals was distinguished in the fraction larger than 300 μm and elutriated one, and weak one was present in the fraction less than 106 μm and between 106 μm and 300 μm. Clear peak of smectite-like minerals was not detected.
In soil B, the peak originated from kaolinite-like minerals was observed in the fractions other than >300 μm. The peak of hornblende-like minerals was observed in the fractions less than 106 μm and between 106 μm and 300 μm. The peak of mica-like minerals was distinguished in the fractions elutriated and between 106 μm and 300 μm, and weak peak was recognized in the other fractions. Peak of smectite-like minerals was distinguished in the elutriated fraction.
In soil C, the peak originated from kaolinite-like minerals was observed in the fractions other than >300 μm. The peak of hornblende-like minerals was observed in the fractions less than 106 μm and between 106 μm and 300 μm. Clear peak of mica-like minerals was not distinguished in any size fractions. Peak of smectite-like minerals was not distinguished in the elutriated fractions, indicating that smectite-like minerals are not present in soil C, even though a weak peak was detected in the fraction of >300 μm.
4. Discussion
Association of fallout radioactive Cs with soils was analyzed by sequential extraction at Norway after Chernobyl accident [8] and at Savannah River site [9]. They indicated that radioactive Cs was tightly associated with soil. They suggested that the tight association resulted from adsorption by micaceous minerals. Our results showed that more than 65% of 137Cs resided in the soil samples after treatment of 1 mole L−1 NH4Cl solution and 1 mole L−1 CH3COOH solution, indicating that more than 60% of radioactive Cs were tightly associated with soils.
The irreversible association site of Cs is defined as association sites from which Cs is not desorbed by 1 mole L−1 KCl solution after the saturation by Cs+ ion [10]. The irreversible association site of the soils collected at Ibaraki, Japan, was reported as more than 1 × 10−7 moleCs/gsoil [10]. Since the mineral composition of the soils of Ibaraki is nearly the same as those of the soils collected in the present study, the irreversible association site is sufficiently high for a radioactive Cs of 105 Bq/kg (equivalent to 2.27 × 10−10 mole/kg). These sufficient irreversible association sites result in the irreversible adsorption fraction more than 60% of the radioactive Cs ().
The fraction of irreversibly associated Cs with the sandy soil of Ibaraki increases with increasing contact time and attains steady state [10]. Longer equilibrating time for sorption of Cs by the soils collected at Sweden Lybia reduced the exchangeable fraction by 1 mole L−1 NH4Cl solution [11,12]. Comans et al. [7] have proposed the following procedure of Cs fixation to the “frayed edges” sites of illite: the initial fast process and the secondary slow process may be caused by a fast diffusion of Cs in the open “frayed edges” of illite and a slow diffusion of Cs in the collapsed “frayed edges” sites of illite. The rate of the slow diffusion was estimated for the sandy soil of Ibaraki, Japan, as 0.009 exp (−4 × 10−5 · t) (s−1) [10], where t is time (s). The rate of the slow diffusion becomes approximately 8.8 × 10−18 (s−1) at 10 days. In the present study, we collected the soil samples at the end of April, suggesting that the slow diffusion process of radioactive Cs was almost completed in the soil samples.
XRD analyses () showed the presence of mica-like minerals in soils A and B. Thus, one of the possible irreversible sorption is caused by fixation at the “frayed edge” of mica-like minerals. The high radioactivity of approximately 106 Bq/kg was determined in the elutriated fraction of soils A and B, resulting from the presence of mica-like minerals in the elutriated one. Mica-like minerals are present in the fractions of 106 μm< >300 μm of soil A and >300 μm of soil B. Sum of the fraction of 137Cs distributed to the above fractions was lower than the irreversibly associated fraction obtained by desorption with 1 mole L−1 KCl solution (). More than 95%, 70%, and 85% of radioactive Cs were associated with the fractions other than the elutriated one of the soils A, B, and C, respectively. These results indicate that some fraction of 137Cs was irreversibly associated with the soils even though mica-like minerals is not present.
Desorption of Cs by 1 mole L−1 KCl solution from standard minerals of kaolinite, feldspar, chlorite, and MnO2 was investigated as a function of concentration of Cs [10], indicating that desorption fraction depends on the concentration of Cs. When the concentration of Cs was 10−9 mole L−1, approximately 30% of adsorbed Cs were not desorbed by 1 mole L−1 KCl solution from these standard minerals. In the contrary, more than 95% Cs were desorbed from these standard minerals at Cs concentrations of 10−5 and 10−4 mole L−1 [10]. Similar desorption behavior of Cs of 10−2 mole L−1 was reported by using the soils sampled at Fukushima [13]. The concentration of the radioactive Cs fallout of 1 × 105 Bq is equivalent to 2.27 × 10−10 mole, suggesting that chemical concentration of the sorbed Cs by the soils is less than 10−9 mole. XRD analysis indicates that minerals of kaolinite-like minerals and feldspar are components of the soils. Thus, the minerals of kaolinite-like minerals and feldspar in the soils probably associate tightly with some fractions of radioactive Cs.
For the remediation of the contaminated soils, the treatment using an appropriate reagent solution is effective. A general candidate for the desorption reagent is 1 mole L−1 NH4Cl solution [14] because the ionic size of Cs+ is similar to NH4 +. Our results indicate that approximately 20% of the radioactive Cs is desorbed even at 60°C, implying that the treatment using 1 mole L−1 NH4Cl solution is not effective for the remediation. On the other hand, acid treatment using 1 mole L−1 H2SO4 is more effective than that of 1 mole L−1 NH4Cl solution. About a half of the radioactive Cs were desorbed from the soils. These results strongly suggest that acid treatment and higher equivalent weight is one of the remediation methods for the contaminated soils. Unfortunately, acid treatment may change the soil property. This indicates that the acid treatment is not recommended to apply directly to the farming lands and residential areas but applies to the “isolated” contaminated soils from residential areas and farming lands. These “isolated” soils contained higher radioactive Cs more than 100,000 Bq/kg and can be treated by an acid solution to decrease radioactivity. Thus, the acid treatment is an effective method to reduce the amounts of highly contaminated soils.
The other treatment is size fractionation of the soils. Our data indicate that elutriated fraction and the size fraction of <106 μm contained approximately 50% of the radioactive Cs resided after the treatment of the 1 mole L−1 NH4Cl and 1 mole L−1 CH3COOH solutions. In Japanese rice paddy, the “shirokaki” treatment, which ploughs and irrigates Japanese rice paddy before transplanting rice seeding, is carried out. The “shirokaki” treatment eliminates small-sized fraction of paddy soil. Thus, the “shirokaki” treatment is effective for the elimination of radioactive Cs.
Our preliminary results were obtained for only three kinds of soils collected in Fukushima prefecture. We should examine the desorption behavior of radioactive Cs from different kinds of the contaminated soils. For the examination of desorption of radioactive Cs, size fractionations and XRD analysis should be accompanied with the desorption experiment using appropriate reagent solutions.
Related Research Data
References
- Chino , M. , Nakayama , H. , Nagai , H. , Terada , H. , Katata , G. and Yamazawa , H. 2011 . Preliminary estimation of release amounts of 131I and 137Cs accidentally discharged from the Fukushima Daiichi nuclear power plant into the atmosphere . J. Nucl. Sci. Technol , 48 : 1129 – 1134 .
- Kawamura , H. , Kobayashi , T. , Furuno , A. , In , T. , Ishikawa , Y. , Nanayama , T. , Shima , S. and Awaji , T. 2011 . Preliminary numerical experiments on oceanic dispersion of 131I and 137Cs discharged into the ocean because of the Fukushima Daiichi nuclear power plant disaster . J. Nucl. Sci. Technol , 48 : 1349 – 1356 .
- Kato , H. , Onda , Y. and Teramage , M. Depth distribution of 137Cs, 134Cs, and 131I in soil profile after Fukushima Daiichi nuclear power plant accident . J. Environ. Radioactivity , in press
- Tanaka , K. , Takahashi , Y. , Sakaguchi , A. , Umeo , M , Hayakawa , S , Tanida , H , Saito , T and Kanai , Y . 2012 . Vertical profiles of iodine-131 and cesium-137 in soils in Fukushima prefecture related to the Fukushima Daiichi nuclear power station accident . Geochem. J , 46 : 73 – 76 .
- Tamura , T. and Jacobs , D.G. 1989 . Structural implication in cesium sorption . Health Phys , 2 : 391 – 398 .
- Sawhney , B.L. 1970 . Potassium and cesium ion selectivity in relation to clay mineral structure . Clay Clay Miner , 18 : 47 – 52 .
- Comans , R.N. , Haller , M. and De Preter , P. 1991 . Sorption of cesium on illite: Non-equilibrium behaviour and reversibility . Geochim. Cosmochim. Acta , 55 : 433 – 440 .
- Riise , G. , Bjornstad , H.E. , Lien , H.N. , Oughton , D.H. and Salbu , B. 1990 . A study on radionuclide . J. Radioanal. Nucl. Chem , 142 : 531 – 538 .
- Knox , A.S. , Hinton , T.G. and Kaplan , D.I. 2001 . Bioavailability of Radioactive Cesium in Old R Discharge Canal, R-Canal, Pond A, and the Adjacent Flood Plain, WSRC-TR-2001-00455 , Virginia : US department Energy .
- Ohnuki , T. 1994 . Sorption characteristics of cesium on sandy soils and their components . Radiochim. Acta , 65 : 75 – 80 .
- Shenber , M.A. and Eriksson , A. 1993 . Exchangeability of caesium in various soils . Sci. Total Environ , 138 : 271 – 279 .
- Evans , D. , Alberts , J.J. and Clark III , R.A. 1983 . Reversible ion-exchange fixation of cesium-137 leaching to mobilization from reservoir sediments . Geochim. Cosmochim. Acta , 47 : 1041 – 1049 .
- Iwata , H. , Shiotsu , H. , Kaneko , M. and Utsunomiya , S. Indiana, USA, in press . “ Nuclear accidents in Fukushima, Japan, and exploration of effective decontaminant for the 137Cs-contaminated soils ” . In Advanced in Nuclear Fuel , Edited by: Revankar , S.T. Purdue University School of Engineering .
- Roed , J. and Anderson , K.G. 1996 . Clean-up of urban areas in the CIS countries contaminated by Chernobyl fallout . J. Environ. Radioactivity , 33 : 107 – 116 .