Abstract
A system for the in situ monitoring of corrosion depth via electrical resistance measurements was applied to study the corrosion rate of type 316L stainless steel at 553 K in pure water. Corrosion depth was measured using a 50 μm diameter wire probe mounted axially in the tube. Measurements were in good agreement with literature data for both the hydrogen water chemistry (HWC) condition and the normal water chemistry (NWC) condition. Oxide film analyses by scanning electron microscopy and laser Raman spectroscopy on the wire probe and the tube showed no effects from shape of the test specimens or the application of electric current. Corrosion kinetics was evaluated by fitting equations to the measurements. Data for the HWC condition could be fitted by a two-step logarithmic–parabolic law. A single-step logarithmic law fitted data for the NWC condition. Changes in corrosion rate by the water chemistry changes were readily detected with the technique. Corrosion depth change could be observed for the water chemistry change from the NWC condition to the HWC condition with electrochemical corrosion potential (ECP) of −0.56 V vs. standard hydrogen electrode, which is lower than the ECP that the phase of iron oxide changes from α-Fe2O3 to Fe3O4.
1. Introduction
The reduction of radiation exposure during plant inspection is an ongoing issue for nuclear power plants [Citation1,Citation2]. One of the main sources of exposure is cobalt activity accumulated on the piping surfaces. Radioactive Co-60 is generated in the core by neutron exposure of non-radioactive Co-59 and radioactive Co-58 is generated by neutron exposure of non-radioactive Ni-58. The radioactive cobalt is transported as ions in water from the core region to the surfaces of piping connected to the reactor pressure vessel (RPV). Then, the ions are incorporated into an oxide film which grows on the piping surfaces [Citation3]. Knowing the corrosion kinetics of the piping material is important for evaluating the activity accumulation and the radiation field build-up.
Type 316L stainless steel (316L SS) is widely used for piping in Japanese and other boiling water reactors (BWRs) [Citation4]. The piping of the primary loop recirculation system (PLR), which is the main source of radiation exposure, is connected to the PLR pump which circulates water in the RPV. Water at high temperature (about 553 K) and of high purity (electrical conductivity less than 30 μS m−1 at room temperature, although with radiolytic oxygen, hydrogen peroxide and hydrogen present) is circulated in the PLR piping. Hydrogen is also intentionally added in some plants to mitigate stress corrosion cracking of structural materials. In the case of no hydrogen addition, oxygen at 100 μg kg−1 and hydrogen peroxide at 200 μg kg−1 are typically contained in the water (called the normal water chemistry (NWC) condition) [Citation5,Citation6]. In the case of hydrogen addition, the hydrogen concentration depends upon the plant specifications and the plant operating characteristics, but a concentration of 100–150 μg kg−1 is typical (called the hydrogen water chemistry (HWC) condition).
The corrosion rate of stainless steel in high-temperature de-oxygenated water has been postulated as being controlled by the diffusion of metal ions across the Cr2O3-based oxide layer, and it conforms to a parabolic rate law [Citation7]. Other reports have expressed total corrosion rate of 316L SS in oxygenated water at 553–563 K as depending upon time raised to the 2/3 power [Citation8] or being controlled by a logarithmic rate law [Citation9]. Corrosion depths at an early time of 200 h are about 50–80% of those at 1000 h [Citation9,Citation10]. So, corrosion rate at time less than 200 h is important for knowing the corrosion kinetics.
The corrosion rate of 316L SS in high-temperature pure water depends upon the water chemistry, which can include some amounts of dissolved oxygen, hydrogen and hydrogen peroxide, as well as the resulting electrochemical corrosion potential (ECP) [Citation10,Citation11]. Corrosion rate was seen to increase, depending on the dissolved oxygen concentration between <5 μg kg−1 and 1000 μg kg−1, in corrosion tests of specimens immersed in 556 K pure water for up to 1000 h [Citation10]. However, the polarization curve in 553 K de-oxygenated pure water was reported to have an active state peak at about −0.36 V vs. standard hydrogen electrode (V vs. SHE) and passive state at about −0.27 V vs. SHE [Citation11].
The characteristics of oxide films formed in high-temperature pure water were also reported to depend on the water chemistry and were expected to affect corrosion [Citation12]. What is more, the effects of hydrogen peroxide on the characteristics of oxide films may be different from those of dissolved oxygen, even though the ECP is the same [Citation13]. It is clearly important to expose specimens to high-temperature water containing hydrogen peroxide to study corrosion rate under the NWC condition.
The characteristics of oxide films formed in high-temperature pure water were also reported to change followed by the water chemistry change from NWC condition to HWC condition and vice versa [Citation12]. Thickness of oxide film asymptotically decreases to about 0.5 μm depending on the water chemistry condition [Citation12]. However, the effects of water chemistry changes on the corrosion depth or the corrosion rate have not been clarified.
In the work reported here, a system for monitoring the corrosion depth in situ by measuring electrical resistance was applied to study the corrosion depth of 316L SS at 553 K in pure water. In situ monitoring of corrosion depth is useful to obtain the corrosion rate of the early stage or the effects of water chemistry change on corrosion rate, because it is not easy to remove a specimen from high-temperature water exposed for hour or less. Corrosion depth of 316L SS in 553 K water was measured under the HWC condition with hydrogen at 150 μg kg−1 and under the NWC conditions with hydrogen peroxide at 200 μg kg−1 and the kinetic expressions for the corrosion were formulated for both conditions. The effects on corrosion depth on water chemistry change from NWC condition to HWC condition and vice versa were also studied. The ECP was also measured to consider the effect of ECP, as well as water chemistry.
2. Experimental procedure
2.1. Measurements of corrosion depth and ECP
The corrosion depth of 316L SS in 553 K pure water has been reported to be of the order of a micrometre or less [Citation8–Citation12]. The corrosion depths of 316L SS wire specimens (diameter 50 μm) were evaluated using electrical resistance measurements applying a constant current (I) and measuring the potential drop (V) intermittently to obtain the resistance (R) via Ohm's law (Equation (1)). The resistance R is also given by the resistivity (ρ), length (l) and radius (r) of the wire as Equation (2), so that r can be obtained from Equation (3). Supposing uniform (general) corrosion, corrosion depth (Δr) is calculated from the initial value (r 0) and the value (rt ) at the immersion time (t) from Equation (4).
The change of R by corrosion is represented by Equation (5) approximately.
Here, R 0 is the initial resistance of the wire probe and Rt is the resistance of the wire probe at time t. Equation (5) indicates that it is desirable for r to be small to increase the change of R.
A schematic drawing of the in situ monitoring system is shown in . The 316L SS wire probe (diameter, 50 ± 5 μm; length, 18 cm; composition [%]: Mn, 1.28; Ni, 10.55; Cr, 17.99; Mo, 2.99; Fe, balance) was mounted axially in the 316L SS tube (inner diameter, 7.04 mm; composition [wt%]: Mn, 1.69; Ni, 12.15; Cr, 18.15; Mo, 2.58; Fe, balance). A photograph of the 316L SS wire probe is also shown in . Lead wires of 316L SS (diameter, 0.5 mm) were silver-soldered to both ends of the wire probe and insulated with polytetrafluoroethylene (PTFE) heat-shrink to avoid contact with water. The 316L SS wire probe and the 316L SS tube were used after degreasing by acetone without polishing. No large scratches were seen on the 316L SS wire probe surfaces as shown in for scanning electron microscopy (SEM) images of the surface and cross section of as-received 316L SS wire probe. Oxide film was not detected on the surfaces of the 316L SS wire probe by laser Raman spectroscopy (LRS).
Figure 1. Schematic of in situ monitoring system. (a) Schematic of test section and measurement systems and (b) photo of wire probe connected with lead wire.
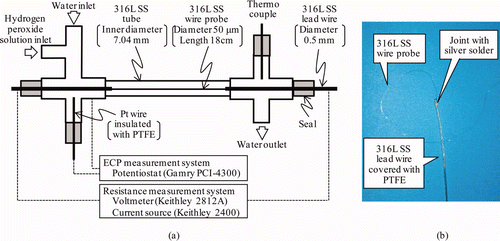
Figure 2. SEM image of (a) cross section and (b) surface of as-received 316L SS wire probe.

A platinum wire insulated with PTFE and a thermocouple were mounted close to the wire probe to measure the ECP and temperature, respectively.
A current source (Keithley 2400) of 1 V with the resolution of 10−9 V and a voltmeter (Keithley 2812A) of 10 mA with the resolution of 5 × 10−6 mA were connected to the 316L SS wire probe. The resistance of the 316L SS wire probe was measured every 2 min. Current should be low to avoid pool boiling on the 316L SS wire probe surfaces. In this work, current of 1 mA was applied. For this measuring condition, the amount of increasing temperature (ΔT ) was calculated to be 0.0023 K from Fourier's law (Equation (6)) using Gnielinski equations for Nusselt number (Nu; Equation (7)), pipe friction coefficient (f, Equation (8)), Reynolds number (Re ), Prandtl number (Pr), thermal conductivity (kf ), equivalent diameter of flow path (d) and thermal flux (q) [Citation14,Citation15].
A potentiostat (Gamry PCI-4300) was connected to the platinum wire and the 316L SS tube and the ECP of the tube was measured every 5 min. Platinum wire was worked as the hydrogen electrode during hydrogen injection of 150 μg kg−1. Potential of the platinum wire (E Pt,HWC ) at 533 K was calculated to be −0.52 V vs. SHE under the HWC condition with hydrogen at 150 μg kg−1 from the Nernst equation (Equation (9)) using Henry's constant of hydrogen (kH) reported in [Citation16], molar fraction of dissolved hydrogen (C H2 ), activity of hydrogen ion ([H+]), gas constant (RG) and temperature (T). The ECP of 316L SS tube (E 316LSS) vs. SHE was calculated from Equation (10) using measured potential of platinum wire (E meas) against 316L SS tube.
Potential of platinum wire is constant under the NWC condition [Citation17]. Potential of platinum wire under the NWC condition (E Pt,NWC) was obtained empirically. Potential of 316L SS tube is controlled by hydrogen peroxide concentration. So, ECP of 316L SS tube under the NWC condition is the same as that under overlapping term of the HWC condition and the NWC condition. On the contrary, platinum wire works as a quasi-hydrogen electrode under the overlapping term of the HWC condition and the NWC condition because the molar ratio of hydrogen to hydrogen peroxide is more than 1 [Citation17,Citation18]. Potential of platinum wire can be calculated from Equation (11) using measured potential of platinum wire against 316L SS tube under the NWC condition (E′meas) and under the overlapping term of the HWC condition and the NWC condition (E′′meas). The ECP of 316L SS tube (E 316LSS) vs. SHE was calculated from Equation (12) with measured potential of platinum wire (E meas) against 316L SS tube under the NWC condition.
2.2. Apparatus and water chemistry conditions
Corrosion depths were measured using the high-temperature, high-pressure loop shown in . Operating conditions are listed in . The water in the pure water tank was circulated to the ion exchange resin column continuously to keep electrical conductivity below 30 μS m−1. For the NWC condition, the water was sparged with argon. For the HWC condition, nitrogen mixed with 10% hydrogen gas was used to control the dissolved hydrogen concentration to 150 μg kg−1. The main water flow rate was kept at 10.8 kg h−1 using a metering pump. Pressure at the test section was set to 10 MPa to avoid boiling and the water was heated to 553 K at the test section using a heat exchanger and electrical heater. After reaching the steady state, temperature was controlled to within ±1K in the test section. Hydrogen peroxide (45 mg kg−1) was injected just upstream from the test section to avoid its decomposition before reaching the test section for getting the NWC condition. The flow rate of hydrogen peroxide solution was set to 0.048 kg h−1 using a metering pump to control its concentration to 200 μg kg−1 at the injection point. After traversing the test section the water was cooled and then deionized by passing through an ion exchange resin.
Figure 3. Outline of high-temperature, high-pressure loop.
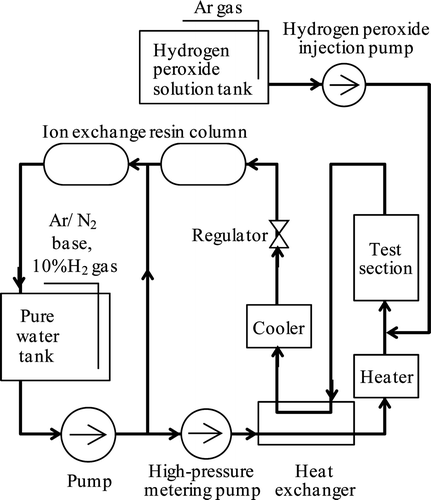
Table 1. Operating conditions of high-temperature, high-pressure loop.
Water chemistry conditions are summarized in . Runs 1 and 2 demonstrated the in situ measurement of corrosion depth via the resistance measurements of the wire probe and provided corrosion kinetics under typical BWR water chemistry conditions. The effects of water chemistry change on corrosion depth were studied in Runs 2 and 3. For the case of Run 2, ECP under the HWC condition was controlled to be lower than the ECP at which the phase of iron oxide changes from α-Fe2O3 to Fe3O4. For the case of Run 3, ECP under the HWC condition was kept higher than the ECP at which the phase of iron oxide changes from α-Fe2O3 to Fe3O4.
Table 2. Water chemistry conditions.
In Run 1, specimens were immersed under the HWC condition for 168 h. In Run 2, the specimens were immersed for 118 h under the NWC condition, 191 h under the HWC condition and then 51 h again under the NWC condition. In Run 3, the specimens were immersed for 120 h under the NWC condition and then 90 h under the HWC condition.
2.3. Oxide film analysis
Surface and cross section morphologies and chemical compositions of oxide films formed on the 316L SS wire probe and 316L SS tube were examined by SEM and LRS, respectively, to consider the effects of shape of the test specimens and current application on the corrosion.
The shape of test specimens will affect the diffusion amount of hydrogen peroxide or hydrogen to the metal surfaces and the diffusion amount of metal ion from metal surfaces. Diffusion layer thickness (δ) was calculated to be 169 μm from Equation (13) using the mass transfer coefficient of straight pipe (Km , Equation (14)), diffusion coefficient of hydrogen peroxide (D), equivalent diameter of flow path (d), Reynolds number (Re) and Schmidt number (Sc) [15,19,20]. Diffusion coefficient of hydrogen peroxide (D) at 553 K was obtained from the Einstein–Stokes equation (Equation (15)) using viscosity of water (η), Boltzmann constant (k b) and radius of chemical species (r C) [Citation15]. Equivalent diameter of flow path (d) was calculated from Equation (16) using volume (M) and surface area (S) of test section.
Diffusion layer thickness is about three times as large as the radius of 316L SS wire probe (25 μm), but which is much smaller than the inner radius of 316L SS tube (3.52 mm). Hydrogen peroxide or hydrogen will diffuse two-dimensionally to the 316L SS wire probe surfaces, but will diffuse one-dimensionally to the 316L SS tube surfaces. Diffusion amount of hydrogen peroxide or hydrogen to the metal surfaces for the 316L SS wire probe will be about eight times as large as that for the 316L SS tube by the calculation using Equation (17).
On the contrary, metal ion concentration dissolved form the 316L SS wire probe surfaces will be eight times as dilute as that dissolved from the 316L SS tube surfaces. Corrosion of the 316L SS wire probe may be larger than that of the 316L SS tube due to these phenomena.
The shape of specimens may also affect the surface morphologies or chemical composition of oxide film. Corrosion increases the volume of 316L SS because the Pilling–Bedworth ratios of iron, nickel and chromium, major elements included in 316L SS, are 2.06, 3.92 and 1.68, respectively [Citation21]. Oxide film thickness is 1 μm or less, which is a significant thickness relative to the 316L SS wire probe radius but is a negligible thickness relative to the 316L SS tube radius. So oxide film on the 316L SS tube surface may grow one-dimensionally. On the contrary, oxide film on the 316L SS wire probe may grow two-dimensionally.
Current application may function as cathodic protection [Citation22,Citation23], which may affect surface and cross section morphologies or chemical composition of oxide film. However, the increased temperature of the 316L SS wire probe by supplying a current is negligible as described in Section 2.1.
3. Results and discussion
3.1. Effects of temperature elevation
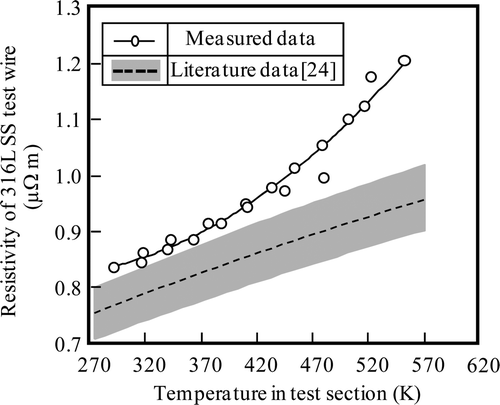
Measurements of resistance during temperature elevation in Run 1 are shown in . Data considered to be for the steady state at each temperature are plotted as open circles. Set temperatures are also shown in . Based on the measurements during temperature elevation, the temperature dependence of specific resistivity of the 316L SS wire probe was obtained and is shown in , along with literature data for 316L SS [Citation14]. Resistivity was calculated supposing that the diameter of 316L SS wire probe was 50 μm. The result was empirically expressed as Equation (18).
Figure 4. Resistance change during temperature elevation for Run 1.
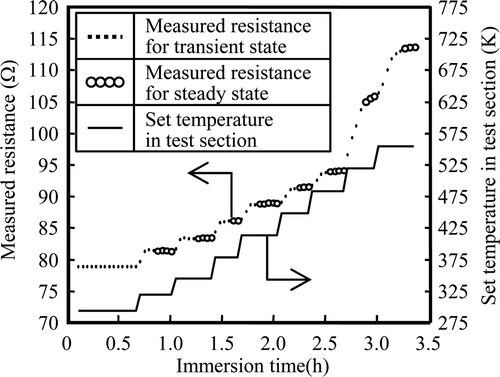
Figure 5. Temperature dependence of resistivity of 316L SS wire probe.
Here, c 1 = 3.434 × 10−12 Ω m K−2, c 2 = −1.502 × 10−9 Ω m K−1, c 3 = 9.843 × 10−7 Ω m.
The correlation coefficient is 0.998, which is better than that for first order fitting (correlation coefficient: 0.967). These results are about 25% higher than the literature data at 553 K. Diametrical variation (±5 μm) of 316L SS wire probe is considered to be one reason for the deviation of ±20%. Some data for annealed specimens have been summarized in the literature [Citation24]. Data scatter of ±6% (grey area of ) is attributed to compositional variation within the specifications of 316L SS and this is a second reason for the resistivity difference. Regarding the effect of cold working on resistivity of 316L SS, it was reported that resistivity of specimens strained 40% in tension is about 1% higher than that of annealed specimens [Citation25]. The effect of heat treatment is considered as a third reason. Resistivity was reported to change about 10–100% in precipitation-hardened type AISI 631 SS [Citation24]. However, for austenitic type AISI 660 SS, resistivity changed less than 5% [Citation24]. So, the effect of heat treatment may be small for 316L SS, because 316L SS is one type of austenitic stainless steel. From these considerations, the compositional variation is considered to be the most probable cause.
3.2. Fluctuation of resistance measurements
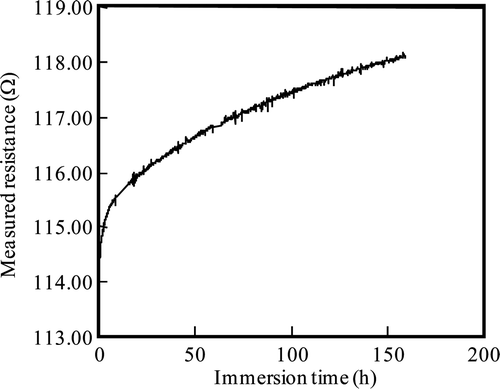
Measurements of resistance at 553 K in Run 1 are shown in . After reaching the set temperature at the test section, the corrosion depth measurement was started and the immersion time was defined as 0 h. Fluctuations in the resistance measurements are within ±0.05 Ω , which corresponds to a corrosion depth of ±0.007 μm. Since corrosion depths are about 0.5 μm, the fluctuations are about ±1.4%.
Figure 6. Immersion time dependence of measured resistance for Run 1.
3.3. Corrosion depth
The measurements of corrosion depth for Run 1 are shown in . Measured ECP at the steady state is −0.56 V vs. SHE (listed in ). Literature data for 561 K water containing small amounts of dissolved oxygen (<5 μg kg−1) with no hydrogen (no ECP reported) [Citation10] and for 561 K water containing dissolved oxygen (15 μg kg−1) and dissolved hydrogen (150 μg kg−1; ECP of −0.46 V vs. SHE) [Citation12] are also plotted. The results of this work are in good agreement with the data of [Citation12], although the data of [Citation10] are about one order of magnitude lower. Since corrosion at or near the activate state is known to be sensitive to ECP, and since a slight amount of oxygen will change the ECP, the deviation from [Citation10] results may be due to differences in ECP. Hydrogen enhances the anodic current, which would cause of lowering the ECP even though oxygen concentration is the same [Citation26]. Hydrogen is the reducing agent to dissolve the protective oxide film and that might enhance the corrosion.
Figure 7. Comparison of corrosion depth of 316L SS under HWC condition among literature data and results of this work for Run 1.
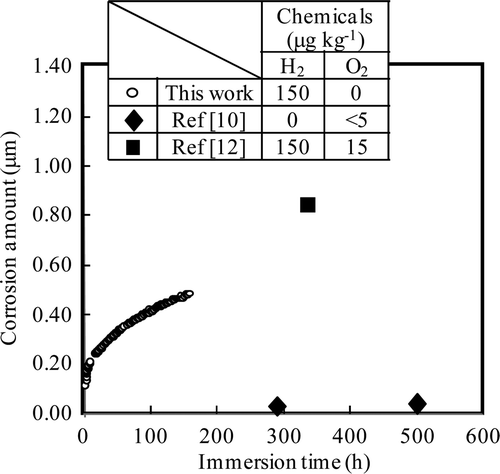
Table 3. Measured ECP of steady state.
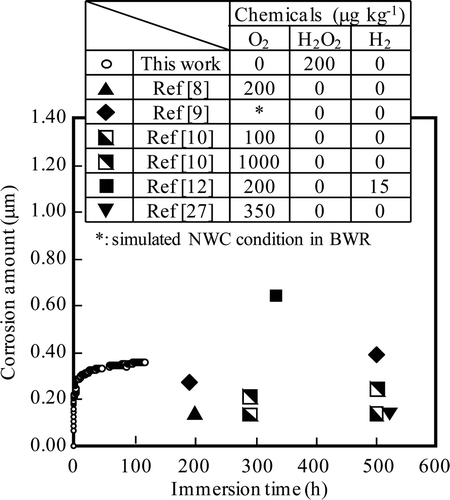
The results of corrosion depth measurements in pure water at 553 K under the NWC condition for 118 h (Run 2) are shown in along with literature data and the experimental conditions at which they were obtained [Citation8–Citation10,Citation12,Citation27]. Measured ECP of the NWC condition (NWC 1) was +0.15 V vs. SHE. The results of this work for water containing hydrogen peroxide are in good agreement with the rather scattered literature data, which were obtained for water containing oxygen.
Figure 8. Comparison of corrosion depth of 316L SS under NWC condition among literature data and results of this work for Run 2.
3.4. Oxide film analysis
The SEM images of the surface and cross section of the oxide film formed on the wire probe and on the tube under the HWC condition as Run 1 are shown in . For both specimens, octahedral crystallites (constituting the outer layer) are scattered on the surface and along the interface at the inner layer of the oxide film. There is no apparent difference in morphology.
Figure 9. SEM image of surface and cross section of oxide film formed under HWC condition for Run 1.

Raman spectra of the oxide films formed on the wire probe and on the tube under the HWC condition (Run 1) are shown in . Raman spectra were measured at three different points for each specimen because NiFe2O4 and α-Fe2O3 are localized as the outer layer [Citation12]. For both specimens, a peak at 680–685 cm−1 can be detected, which is attributed to FeCr2O4 or Fe3O4. In the case of the wire probe, a peak is also detected at 811 cm−1, and since this peak is also seen on the specimen before exposure in the loop, it cannot be attributed to a compound formed by immersion in high-temperature water.
Kim [Citation12] describes the characteristics of the oxide film formed in 561 K water containing 150 μg kg−1 of hydrogen and 15 μg kg−1 of oxygen as analysed by SEM and transmission electron microscopy (TEM). Large crystallites of γ-Fe2O3 and α-Fe2O3 as the outer layer and fine grains containing FexCr3−x O4 as the inner layer were observed. γ-Fe2O3 and α-Fe2O3 were not detected in the present work. The differences might be due to a slight difference in water chemistry. Only hydrogen (150 μg kg−1) was added and ECP was about −0.56 V vs. SHE in the present work, whereas in [Citation12] oxygen was added to the amount of 15 μg kg−1 and ECP was about −0.46 V vs. SHE. According to thermodynamic calculations [Citation28], the phase of iron oxide changes from α-Fe2O3 to Fe3O4 at an ECP of −0.47 V vs. SHE. Oxygen of 15 μg kg−1 would make the water chemistry of [Citation12] study a little more oxidative than that in the present work, possibly leading to the formation of the higher-order iron oxide.
The SEM images of the surface and cross section of oxide film formed on the wire probe and on the tube after Run 2 are shown in . The specimens were immersed for 118 h under the NWC condition (measured ECP + 0.15 V vs. SHE), 191 h under the HWC condition (measured ECP − 0.56 V vs. SHE) and then 51 h again under the NWC condition (measured ECP + 0.00 V vs. SHE). For both specimens, octahedral crystallites form a densely packed outer layer but no crystallites can be seen for the inner layer. There is no apparent difference in morphology.
Figure 10. Raman spectra of oxide film formed under HWC condition for Run 1.
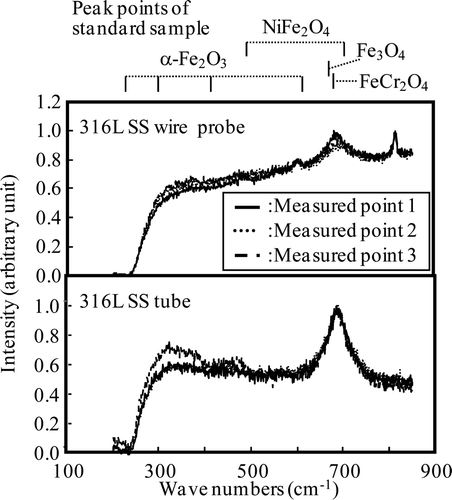
Figure 11. SEM image of surface and cross section of oxide film formed under NWC condition for Run 2.

Raman spectra of the oxide films formed on the wire probe and on the tube after Run 2 are shown in . Raman spectra were also measured at three different points for each specimen. For both specimens, peaks due to α-Fe2O3 (293, 412, 612 cm−1), Fe3O4 (660 cm−1) and NiFe2O4 (497, 699 cm−1) are detected. However, relative peak intensity of α-Fe2O3 for 316L SS tube is smaller than that for 316L SS wire probe. The Raman scattering power of α-Fe2O3 is well known to be much larger than that of Fe3O4 [Citation29]. So, a small difference in the α-Fe2O3 formation under the NWC condition may cause the difference in relative peak intensity of α-Fe2O3.
Figure 12. Raman spectra of oxide film formed under NWC condition for Run 2.
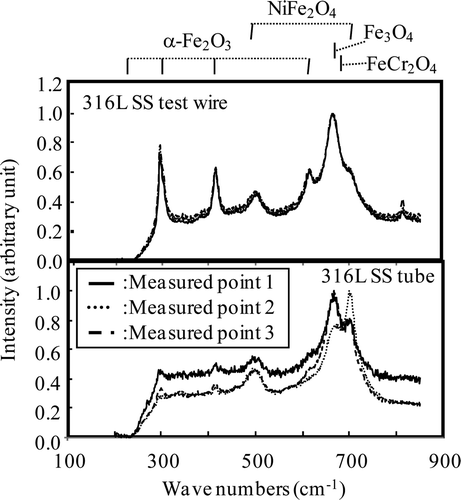
Again, in the case of the wire probe, the peak at 811 cm−1 is attributed to contamination before exposure in the loop. Kim [Citation12] reported on the SEM and TEM characterization of oxide films formed in 561 K water containing nominally 15 μg kg−1 of hydrogen and 200 μg kg−1 of oxygen with some changes in conditions. Specimens were immersed under the NWC condition for 2 weeks, the HWC condition for 2 weeks and then the NWC condition again for 2 weeks. As in the experiments reported here, large crystallites of γ-Fe2O3, α-Fe2O3 and NiFe2O4 as the outer layer and fine grains containing FexCr3−x O4 as the inner layer were detected.
The results of corrosion measurements and oxide film analyses on the wire probe and the tube show no effects from shape of the test specimens or the application of electric current. Evaluating corrosion depth under high-temperature pure water by in situ measurements of the resistance of a thin wire is therefore judged to be a useful and valid technique.
3.5. Corrosion kinetics
Corrosion of stainless steel in pure water at 553 K is thought to be controlled by a parabolic rate law or a logarithmic rate law described as Equation (19) or Equation (20), respectively [Citation7,Citation9]. Consequently, the corrosion depth data were fitted with the equations:
The fitting results of the corrosion depth measured under the HWC condition are shown in . Neither of the two equations can be fitted individually. Corrosion is therefore postulated to be controlled by a logarithmic rate law for the initial 1.4 h and by a parabolic rate law thereafter. Thus, data were fitted by Equation (20) until 1.4 h and data after 1.4 h were fitted by Equation (21).
Figure 13. Fitting results of corrosion depth under HWC condition for Run 1.
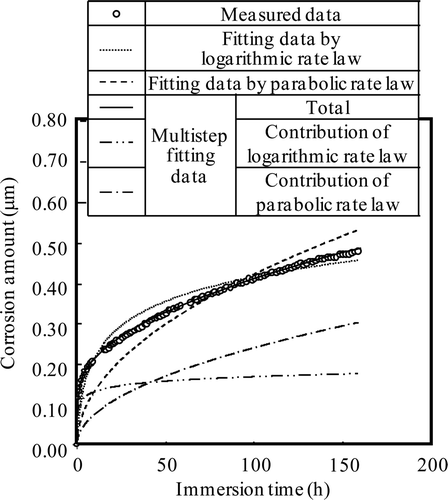
Here, W 0 is the corrosion depth at 1.4 h and t 0 is the period of data fitted by the logarithmic rate law (1.4 h). This multi-step regression curve fits the measured data well; the logarithmic rate law until 1.4 h with b′ = 115.7 h−1 and k = 0.02665 μm h−1 and the parabolic rate law after 1.4 h with a′ = 0.02651 μm h−0.5. Such multi-step control of corrosion may reflect the change in kinetics as formation of the oxide film on the bare surface gives way to metal ion diffusion through the film.
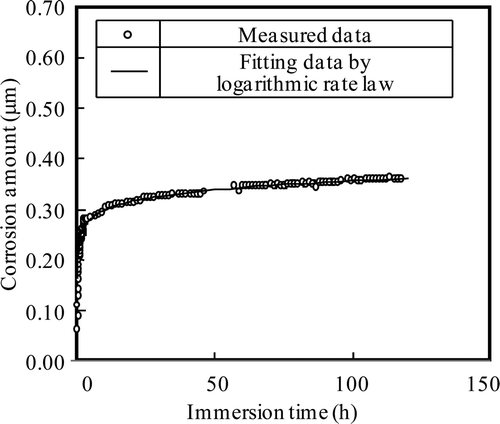
The fitting results of the corrosion depth measured under the NWC condition (NWC 1) in Run 2 are shown in . In this case, the data are fitted nicely by the logarithmic rate law (b′ = 3191 h−1 and k = 0.02834 μm h−1).
Figure 14. Fitting results of corrosion depth under NWC condition for Run 2.
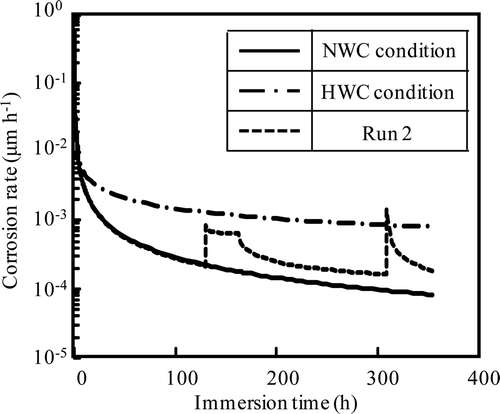
Corrosion rates were calculated using the fitting equation for the HWC condition (Run 1) and the NWC condition (Run 2), and are shown in . Corrosion rate of the HWC condition is larger than that of the NWC condition excluding the initial period of less than 0.1 h. The active state at low ECP (−0.34 V vs. SHE) was observed by anodic polarization curve measurement and the critical current of passivation was reported to be 28 mA m−2 [Citation11]. Corrosion rate was calculated from the critical current of passivation as about 3.7 × 10−3 μm h−1 supposing corrosion by divalent metal ions and density of 79.8 t m−3 and the present result of the present work (1.5 × 10−3 μm h−1 at 100 h for the HWC condition) is in good agreement. Passive current density was also reported to be 5 mA m−2 [Citation11]. Corrosion rate calculated from passive current density was 6.7 × 10−4 μm h−1, with which the result of the present work is in good agreement (3.7 × 10−4 μm h−1 at 100 h for the NWC condition). However, transpassivation potential was 0.00 V vs. SHE, which is lower than the ECP for the NWC condition in the present work. Corrosion rate increases with increasing ECP above transpassivation potential. This may imply that not only ECP but also hydrogen peroxide itself affect the corrosion rate. Hydrogen peroxide may assist the oxide formation to control corrosion.
Figure 15. Corrosion rate under HWC condition, NWC condition and Run 2 calculated using fitting equations.
Corrosion kinetics and corrosion rate clearly depend on water chemistry such as ECP and hydrogen peroxide. Differences in corrosion kinetics and corrosion rate may contribute to the difference in the oxide film. Base metal is covered by the oxide film under the NWC condition, which controls the corrosion rate. However, base metal is covered insufficiently by the oxide film under the HWC condition.
3.6. Effects of water chemistry change on corrosion
Corrosion depths for Run 2 and Run 3 are shown in and , respectively. For the case of Run 2, ECPs of the first NWC condition (NWC 1), the HWC condition and the second NWC condition (NWC 2) were +0.15, −0.56 and +0.00 V vs. SHE at the steady state, respectively. Water chemistry was changed from the NWC condition to the HWC condition at 118 h. After the water chemistry change, 14 h was needed to get the steady state of ECP. Water chemistry was changed from the HWC condition to the NWC condition at 309 h. After the water chemistry change, 12 h was needed to get the steady state of ECP. The change from the NWC condition to the HWC condition increases the corrosion depth above the extrapolated line to continue corrosion under the NWC condition at the same time as ECP is decreased to −0.56 V vs. SHE. By contrast, the change from the HWC condition to the NWC condition at 309 h immediately increases the corrosion depth above the extrapolated line for corrosion under the HWC condition after the chemistry change.
Figure 16. Effects of water chemistry change on corrosion depth for Run 2.
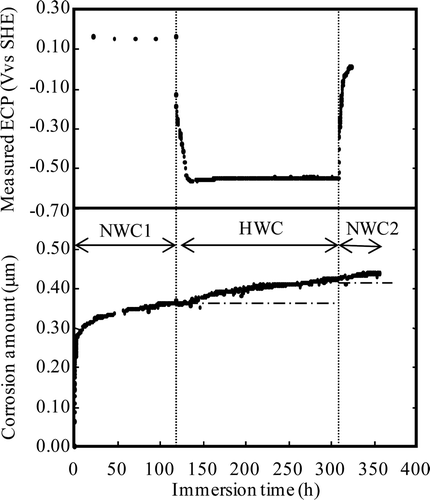
Figure 17. Effects of water chemistry change on corrosion depth for Run 3.
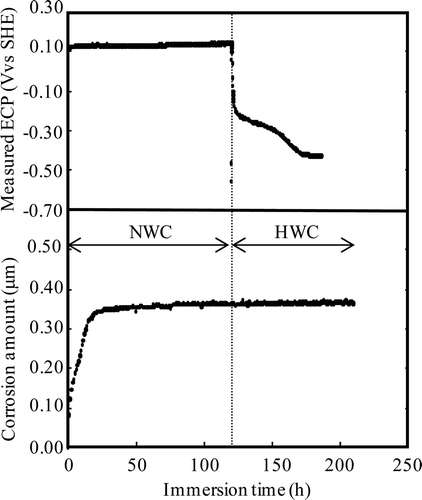
For the case of Run 3, ECPs of NWC and HWC were +0.15 and −0.42 V vs. SHE, respectively. Water chemistry was changed from the NWC condition to the HWC condition at 118 h. After the water chemistry change, 54 h was needed to get the steady state of ECP. The ECP of the HWC condition for Run 3 is higher than that for Run 2. The reason for the ECP difference itself and for the difference in the decrease of ECP between Run 2 and Run 3 under HWC condition may be because of a small amount of oxygen ingress into the pure water tank in Run 3. Deionized water sparged by argon was supplied continuously during Run 3. Oxygen might not have been sufficiently degassed from the water. The change from the NWC condition to the HWC condition did not increase the corrosion depth for Run 3.
The difference of corrosion behaviour by the water chemistry change might be attributable to ECP. According to thermodynamic calculations [Citation28], the phase of iron oxide changes from α-Fe2O3 to Fe3O4 at an ECP of −0.47 V vs. SHE. Since in Run 2 the ECP decreases below −0.47 V vs. SHE under the HWC condition, the oxide film can undergo reconstruction and promote an increase in corrosion.
Corrosion depth was fitted by the Equation (22) and corrosion rates were calculated.
Here, a′ = 0.008 μm h0.05, t′ = 130 h, W′ = 0.364μm and n = 0.95 from 130 h to 160 h; a′ = 0.0012 μm h0.32, t′ = 160 h, W′ = 0.384 μm and n = 0.68 from 160 h to 309 h; and a′ = 0.0019 μm h0.45, t′ = 309 h, W′ = 0.423 μm and n = 0.55 from 309 h to 360 h. The results are shown in . Corrosion rates under the HWC condition in Run 2 (from 130 h to 309 h) are lower than that for the HWC condition measured in Run 1. This means that oxide film formed under NWC condition until 118 h provided corrosion protection. However, oxide film for corrosion protection may be partially damaged, according to the results that the increase of corrosion rate only occurred when ECP is below −0.47 V vs. SHE at which the phase of iron oxide changes from α-Fe2O3 to Fe3O4.
4. Conclusion
The in situ system for monitoring corrosion via electrical resistance measurements of a thin wire was applied to study the corrosion of 316L SS in pure water at 553 K. Corrosion depths were measured under the HWC condition (150 μg kg−1 of hydrogen) and the NWC condition (200 μg kg−1 of hydrogen peroxide) and the kinetic expressions for the corrosion were formulated for both conditions. The effects on corrosion depth from water chemistry change from the NWC condition to the HWC condition and vice versa were also studied.
Corrosion depth could be measured using a 50 μm diameter wire probe mounted axially in the 316L SS tube. Measurements were in good agreement with literature data for both the HWC and the NWC conditions. Differences in oxide morphology could not be observed between the 316L SS wire and a 316L SS tube by SEM analysis or in oxide chemical composition between the wire and tube by LRS analysis. The results of corrosion measurements and oxide film analyses on the wire probe and the tube showed no effects from shape of the test specimens or the application of electric current.
Corrosion kinetics was evaluated by fitting equations to the measurements of corrosion depth. The data for the HWC condition could not be fitted individually by either a parabolic or a logarithmic rate law equation, but a two-step logarithmic–parabolic law fitted them very well. A single-step logarithmic law fitted the data for the NWC condition very well.
Changes in corrosion rate by the water chemistry changes were readily detected with the resistance technique. Corrosion amount change could be clearly observed for the water chemistry change from the NWC condition to the HWC condition with ECP of −0.56 V vs. SHE, which was lower than the ECP at which the phase of iron oxide changes from α-Fe2O3 to Fe3O4 (−0.47 V vs. SHE). However, the corrosion amount change could not be observed for the water chemistry change from the NWC condition to the HWC condition with ECP of −0.42 V vs. SHE, which was higher than the phase of iron oxide changes from α-Fe2O3 to Fe3O4.
Abbreviations and nomenclature
| 304 SS: | = |
type 304 L stainless steel |
| 316L SS: | = |
type 316L stainless steel |
| BWR: | = |
boiling water reactor |
| ECP: | = |
electrochemical corrosion potential |
| HWC: | = |
hydrogen water chemistry |
| LRS: | = |
laser Raman spectroscopy |
| NPP: | = |
nuclear power plant |
| NWC: | = |
normal water chemistry |
| PLR: | = |
primary loop recirculation system |
| PTFE: | = |
polytetrafluoroethylene |
| V vs. SHE: | = |
volt versus standard hydrogen electrode |
| RPV: | = |
reactor pressure vessel |
| SEM: | = |
scanning electron microscopy |
| TEM: | = |
transmission electron microscopy |
| a: | = |
constant |
| a′: | = |
constant |
| a″: | = |
constant |
| b: | = |
constant |
| b′: | = |
constant |
| c1: | = |
constant |
| c2: | = |
constant |
| c3: | = |
constant |
| C H2 : | = |
molar fraction of dissolved hydrogen |
| d: | = |
equivalent diameter of flow path |
| D: | = |
diffusion coefficient of hydrogen peroxide |
| E Pt,NWC: | = |
potential of platinum wire under NWC condition |
| E Pt,HWC: | = |
potential of platinum wire under HWC condition |
| E 316LSS: | = |
ECP of 316L SS tube |
| E meas: | = |
measured potential of platinum wire |
| E′meas: | = |
measured potential of platinum wire against 316L SS tube under NWC condition |
| E′′meas: | = |
measured potential of platinum wire under overlapping term of HWC and NWC conditions |
| f: | = |
pipe friction coefficient |
| [H+]: | = |
activity of hydrogen ion |
| I: | = |
current |
| k: | = |
constant |
| kb: | = |
Boltzmann constant |
| kH: | = |
Henry's constant |
| K m: | = |
mass transfer coefficient of straight pipe |
| l: | = |
length of 316L SS wire probe |
| M: | = |
volume of test section |
| n: | = |
constant |
| Nu: | = |
Nusselt number |
| Pr: | = |
Prandtl number |
| R: | = |
resistance |
| RG: | = |
gas constant |
| R: | = |
radius of 316L SS wire probe |
| rt : | = |
initial radius of 316L SS wire probe |
| r C: | = |
radius of chemical species |
| Re: | = |
Reynolds number |
| R 0: | = |
initial resistance of 316L SS wire probe |
| Rt : | = |
resistance of 316L SS wire probe at t |
| S: | = |
surface area of test section |
| Sc: | = |
Schmidt number |
| t: | = |
immersion time |
| t′: | = |
immersion time from corrosion rate change |
| T: | = |
temperature |
| t 0: | = |
period of data fitted by logarithmic rate law (1.4 h) |
| V: | = |
potential drop |
| W: | = |
corrosion depth |
| W′: | = |
corrosion depth just before corrosion rate change |
| W 0: | = |
corrosion depth at 1.4 h |
| Δr: | = |
corrosion depth of 316L SS wire probe |
| δ: | = |
diffusion layer thickness |
| η: | = |
viscosity of water |
| ρ : | = |
resistivity of 316L SS wire probe |
Acknowledgements
The authors are grateful to A. Feicht, L. Liu and P. Srisukvatananan, research staff in the Nuclear Engineering Group at the University of New Brunswick, for their help with the experiments and the surface analyses. The Natural Sciences and Engineering Research Council of Canada and the CANDU Owners Group are thanked for financial support.
References
- Shaw , R.A. 1979 . Getting at the source: reducing radiation fields . Nucl. Technol , 44 : 97 – 103 .
- Lin , C.C. 2009 . A review of corrosion product transport and radiation field buildup in boiling water reactors . Prog. Nucl. Energy , 51 : 207 – 224 .
- Lister , D.H. 1976 . The transport of radioactive corrosion products in high-temperature water . Nucl. Sci. Eng , 59 : 406 – 426 .
- Okamura , Y. , Sakashita , A. , Fukuda , T. , Yamashita , H. and Futami , T. 2003 . Latest SCC Issue of Core Shroud and Recirculation Piping in Japanese BWRs . Transactions of the 17th International Conference on Structural Mechanics in Reactor Technology . August 17–22 2003 . Paper Number WG01-1
- Wada , Y. , Shigeneka , N. , Uetake , N. and Uchida , S. 1997 . Numerical Simulation of SCC Environment in a BWR Primary Coolant System . Proceedings of the Eighth International Symposium on Environmental Degradation of Materials in Nuclear Power Systems – Water Reactor Vol. 1 . August 10–14 1997 . pp. 574 – 581 .
- Cowan , R.L. , Indig , M.E. , Kass , J.N. , Law , R.J. and Sundberg , L.L. 1986 . Experience with Hydrogen Water Chemistry in Boiling Water Reactors . Water Chemistry of Nuclear Reactor Systems 4 Vol. 1 . October 13–17 1986 . pp. 29 – 36 .
- Robertson , J. 1991 . The mechanism of high temperature aqueous corrosion of stainless steels . Corros. Sci , 32 : 443 – 465 .
- Degueldre , C. , O'Prey , S. and Francioni , W. 1996 . An in-line diffuse reflection spectroscopy study of the oxidation of stainless steel under boiling water reactor conditions . Corros. Sci , 38 : 1763 – 1782 .
- Inagaki , H. , Nishikawa , A. , Sugita , Y. and Tsuji , T. 2003 . Synergy effect of simultaneous zinc and nickel addition on cobalt deposition onto stainless steel in oxygenated high temperature water . J. Nucl. Sci. Technol , 40 : 143 – 152 .
- Ohashi , K. , Honda , T. , Kashimura , E. and Furutani , Y. 1988 . Effect of dissolved oxygen on corrosion of ferrous materials in high temperature and high pure water . Boshoku Gijyutsu (Corros. Eng.) , 37 : 198 – 204 . [in Japanese]
- Tachibana , M. , Ishida , K. , Wada , Y. , Shimizu , R. , Ota , N. and Hara , N. 2012 . Determining factor for anodic polarization curve of typical structural materials of boiling water reactors in high temperature–high purity water . J. Nucl. Sci. Technol , 49 : 253 – 262 .
- Kim , Y.J. 1995 . Characterization of the oxide film formed on type 316 stainless steel in 288°C in cyclic normal and hydrogen water chemistry . Corrosion , 51 : 849 – 860 .
- Miyazawa , T. , Uchida , S. , Satoh , T. , Morishima , Y. , Hirose , T. , Satoh , Y. , Iimura , K. , Wada , Y. , Hosokawa , H. and N. 2005 . Usui, Effects of hydrogen peroxide on corrosion of stainless steel, (IV) Determination of oxide film properties with multilateral surface analyses . J. Nucl. Sci. Technol , 42 : 233 – 241 .
- The Japan Society of Mechanical Engineers . 1986 . JSME Data Book: Heat Transfer , 4th ed. , 55 Tokyo : Maruzen Co., Ltd . [in Japanese]
- The Japan Society of Mechanical Engineers . 1999 . JSME Steam Table , 29 Tokyo : The Japan Society of Mechanical Engineers . [in Japanese]
- Pray , H.A. , Schweickert , C.E. and Minnich , B.H. 1952 . Solubility of hydrogen, oxygen, nitrogen, and helium in water . Ind. Eng. Chem , 44 : 1146 – 1151 .
- Kim , Y.J. 2005 . Effect of variations in simulated BWR water chemistry on high temperature electrochemistry of stainless steel . Corrosion 2005 . April 3–7 2005 . PN05579
- Kim , Y.J. and Andresen , P.L. 2001 . Data Quality, Issues and Guidelines for ECP Measurement in High Temperature Water . Corrosion 2001 . March 11–16 2001 . PN01137
- Berger , F.P. and Hau , K.-F.F.-L. 1997 . Mass transfer in turbulent pipe flow measured by the electrochemical method . Int. J. Heat Mass Transf , 20 : 1185
- Elliot , A.J. , McCracken , D.R. , Buxton , G.V. and Wood , N.D. 1990 . Estimation of rate constant for near-diffusion-controlled reactions in water at high temperatures . J. Chem. Soc. Faraday Trans , 86 : 1539 – 1547 .
- Pilling , N.B. and Bedworth , R.E. 1923 . The oxidation of metals at high temperature . J. Inst. Met , 29 : 529 – 591 .
- Mears , R.B. and Brown , R.H. 1938 . A theory of cathodic protection . Trans. Electrochem. Soc , 74 : 519 – 531 .
- Brown , R.H. and Mears , R.B. 1942 . Cathodic protection . Trans. Electrochem. Soc , 81 : 455 – 483 .
- Bogaard , R.H. , Desai , P.D. , Li , H.H. and Ho , C.Y. 1993 . Thermophysical properties of stainless steels . Thermochim. Acta , 218 : 373 – 393 .
- Yang , S.W. and Spruiell , J.E. 1982 . Cold-worked state and annealing behavior of austenitic stainless steel . J. Mater. Sci , 17 : 677 – 690 .
- Kim , Y.J. 1997 . Electrochemical Interactions of Hydrogen, Oxygen and Hydrogen Peroxide on Metal Surfaces in High Temperature, High Purity Water . Proceedings of the Eighth International Symposium on Environmental Degradation of Materials in Nuclear Power Systems – Water Reactors Vol. 2 . August 10–14 1997 . pp. 641 – 648 .
- Lister , D.H. and Venkateswaran , G. 1999 . Effects of magnesium and zinc additives on corrosion and cobalt contamination of stainless steels in simulated BWR coolant . Nucl. Technol , 125 : 316 – 331 .
- Cubicciotti , D. 1989 . Equilibrium chemistry of nitrogen and potential – pH diagrams for the Fe–Cr–H2O system in BWR water . J. Nucl. Mater , 167 : 241 – 248 .
- Shebanova , O.N. and Lazor , P. 2003 . Raman spectroscopic study of magnetite (FeFe2O4): a new assignment for the vibrational spectrum . J. Solid State Chem , 174 : 424 – 430 .