
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
To determine the equilibrium constant for ferroselite (FeSe2(cr)) dissolution reaction, FeSe2(cr) solubility experiments were performed at 298 ± 1 K from both the over- and under-saturation directions with Fe–Se precipitates that were aged at 348 K. X-ray diffraction (XRD) analysis detected only FeSe2(cr) as the Se solid phase in the equilibrated precipitates. The Eh values of the equilibrated suspensions ranged from −188.6 to −4.9 mV vs. standard hydrogen electrode (SHE) and the pH values ranged from 6.00 to 8.76. Based on the available thermodynamic data, Se42− and Fe2+ are thermodynamically stable within this Eh–pH range. Agreement between the solubility data obtained from the over- and under-saturation directions lends credence to the attainment of equilibrium at 298 ± 1 K. The thermodynamic interpretations using the specific ion interaction theory (SIT) model showed that Eh values and the concentrations of Se and Fe are well represented by the 2FeSe2(cr) solubility reaction (2FeSe2(cr) ⇌ 2Fe2+ + Se42− + 2e−) with log10K○ = −17.09 ± 0.28. The obtained log10K○ value falls within the uncertainty limits of the log10K○ value calculated from the available thermodynamic data.
1. Introduction
Selenium (Se) is one of the key elements in the safety assessment of the geological disposal system because 79Se is a long-lived fission product. The retardation of Se migration by sorption onto the buffer material and host rock is not expected due to the anionic nature of the Se species. Because of slow diffusional transport and nearly nonexistent sorption, it is expected that the solubility phenomenon will control Se concentrations in repository environments. Therefore, accurate information on solubility of Se is required to predict the migration of Se. The oxidation–reduction potential (Eh)–pH diagram of , which is based on thermodynamic database reviewed and compiled by Kitamura et al. [Citation1], illustrates the stability ranges of the dominant Se oxidation states and the predominant solution speciation in aqueous systems with 1 × 10−2 mol dm−3 iron (Fe). Fe was added in this diagram because Fe2+ derived from the corrosion of a carbon steel overpack is assumed to dissolve into groundwater in repository environments. The Eh–pH diagram of is different from the Eh–pH diagram constructed by Iida et al. [Citation2] because Fe was not added when constructing the latter. The Eh–pH diagram of suggests that ferroselite (FeSe2(cr)) is thermodynamically stable under reducing conditions. Because the formation of FeSe2(cr) is probably possible under reducing repository environments, the FeSe2(cr) solubility is of paramount importance in the safety assessment of the geological disposal system. Although there have been several previous studies of Se solubility in the presence of Fe under reducing conditions [Citation3–6], no studies wherein the FeSe2(cr) solubility was measured are reported. Direct evidence of a solubility limit by FeSe2(cr) under reducing conditions was lacking. The following equilibrium constant for FeSe2(cr) dissolution reaction was used to construct the Eh–pH diagram of
(1)
(1)
Figure 1. Eh–pH diagram for Se based on the thermodynamic data provided by Kitamura et al. [Citation1]. The total concentrations of Se and Fe are 1 × 10−5 and 1 × 10−2 mol dm−3, respectively.
![Figure 1. Eh–pH diagram for Se based on the thermodynamic data provided by Kitamura et al. [Citation1]. The total concentrations of Se and Fe are 1 × 10−5 and 1 × 10−2 mol dm−3, respectively.](/cms/asset/d3c99131-6403-4951-b326-8c9be1ab1b7d/tnst_a_1137245_f0001_b.gif)
The above equilibrium constant is a tentative value calculated from the following standard molar Gibbs energy of formation for FeSe2(cr) (ΔfG○m(FeSe2(cr))) [Citation1,Citation7]:
(2)
(2) where ΔfH○m(FeSe2(cr)) is the standard molar enthalpy of formation of FeSe2(cr) and S○m(FeSe2(cr)) is the standard molar entropy of FeSe2(cr). It should be noted that the large uncertainty in ΔfH○m(FeSe2(cr)) leads to a large uncertainty in the calculated log10K○1 value. Considerable uncertainty has surrounded FeSe2(cr) solubility. For these reasons, the objective of this study is to determine the equilibrium constant for FeSe2(cr) dissolution reaction.
Measurement of the Se and Fe concentrations controlled by FeSe2(cr) is required to achieve this objective. Although Doi et al. [Citation3] performed solubility experiments of Se co-existing with Fe under reducing conditions, the Se concentration in 0.45 μm filtrates from 2 months equilibrated suspensions produced from the over-saturation direction was lower than the detection limit of inductively coupled plasma mass spectrometry (ICP-MS). Doi et al. [Citation3] reported that the Se and Fe concentrations were likely to be controlled by the dissolution reaction in EquationEquation (1)(1)
(1) . It can be seen from EquationEquation (1)
(1)
(1) that as the ae- value decreases (i.e., the Eh rises), the concentrations of Se and/or Fe in solution controlled by the dissolution reaction in EquationEquation (1)
(1)
(1) decrease. Lower Eh is required to measure the Se and Fe concentrations controlled by the dissolution reaction in EquationEquation (1)
(1)
(1) . There was a possibility that the rise of the Eh values of the samples used in Doi et al. [Citation3] would result in the undetectable Se concentration. The experiments reported here were designed to make the Se concentration higher than the detection limit of ICP-MS. The Eh–pH diagram of shows the prediction of Se aqueous species. Stability fields of Se solids and Se gas species occupy the range of reducing conditions in an Eh–pH diagram for Se based on the above-mentioned thermodynamic database. In the preparation for the Eh–pH diagram of , the thermodynamic data of Se solids and Se gas species were removed from the above-mentioned thermodynamic database, thereby making stability fields of Se aqueous species visible. As suggested in , Se42− is the predominant solution species present under moderately reduced and neutral pH conditions. FeSe2(cr) dissolution reaction with Se42− and Fe2+ can be described as
(3)
(3)
Figure 2. Eh–pH diagram for the prediction of Se aqueous species based on the thermodynamic data provided by Kitamura et al. [Citation1]. The total concentration of Se is 1 × 10−7 mol dm−3. The experimental data obtained from the over- and under-saturation directions are plotted as open circles and solid symbols, respectively.
![Figure 2. Eh–pH diagram for the prediction of Se aqueous species based on the thermodynamic data provided by Kitamura et al. [Citation1]. The total concentration of Se is 1 × 10−7 mol dm−3. The experimental data obtained from the over- and under-saturation directions are plotted as open circles and solid symbols, respectively.](/cms/asset/60235d0e-f129-404b-ab94-726122379509/tnst_a_1137245_f0002_b.gif)
The K○2 value of the dissolution reaction in EquationEquation (3)(3)
(3) is given by
(4)
(4)
In contrast to the dissolution reaction in EquationEquation (1)(1)
(1) , it can be seen from EquationEquation (4)
(4)
(4) that the higher the Eh value, the higher the concentrations of Se and/or Fe in solution controlled by the dissolution reaction in EquationEquation (3)
(3)
(3) . Where Se42− and Fe2+ are the predominant solution species, there is no need to maintain strongly reduced conditions for the purpose of measuring the Se and Fe concentrations controlled by FeSe2(cr). Therefore, to study FeSe2(cr) dissolution behavior (EquationEquation (3)
(3)
(3) ), moderately reduced and neutral pH conditions were maintained in this study. Kitamura et al. [Citation4] reported that the Fe concentrations were lower than the detection limit of ICP-MS because of alkaline pH. The neutral pH conditions are also preferred for the measurement of the Fe concentration.
In the previous solubility experiments of Se co-existing with Fe, FeSe2(cr) was not detected by X-ray diffraction (XRD) analysis in the equilibrated precipitates produced from the over-saturation direction at room temperature [Citation5], but the transformation from Se(cr) to FeSe2(cr) with increasing experimental run times occurred at 353 K [Citation3]. Also, FeSe2(cr) has been synthesized at a temperature of 353 K [Citation8]. In this study, the precipitate was aged at 348 K because elevated temperature such as that used in this study is expected to promote the formation of FeSe2(cr) and allow deep underground temperature to be realistically simulated.
The ionic strengths of the samples used in the previous solubility experiments [Citation3–Citation6] were relatively low. Determining the solubility-controlling solid of Se at high ionic strength is required to understand Se migration in case of intrusion of saline groundwater for coastal repositories. Therefore, experiments were carried out in the same ionic strength solutions as simulated saline groundwater [Citation9].
2. Experimental
2.1. Solubility experiments
All experiments except ICP-MS (PerkinElmer Japan Co., Ltd. NexION 300) analysis were conducted in a glovebox under a nitrogen atmosphere (O2 < 1 ppm).
Three grams of metallic Se (Wako Pure Chemical Industries, Ltd. 190-00292) were washed in 0.01 mol dm−3 sodium hydroxide (NaOH) (KANTO KAGAKU 37854-08) to remove soluble impurities such as SeO2. The washed metallic Se was dissolved at 353 K in 14 mol dm−3 NaOH produced from mixing 48% NaOH (KANTO KAGAKU 37959-00) and distilled water. This solution was filtered through 0.45 μm membrane filters (ADVANTEC 25HP045AN) to remove the solid material.
To remove organic substances, 7 g of metallic Fe (Mitsuwa Chemical Co., Ltd 53496) were washed three times with 0.05 mol dm−3 hydrochloric acid (HCl) (KANTO KAGAKU 18598-08) followed by washing with distilled water. The washed metallic Fe was dissolved in 2 mol dm−3 HCl (KANTO KAGAKU 18590-08). This solution was filtered through 0.45 μm membrane filters to remove the unreacted metallic Fe.
In order to approach solubility from the over-saturation direction, aliquots of Se and Fe filtrates were added into deionized water to reach initial concentrations of 0.011 mol dm−3 Se and 0.012 mol dm−3 Fe. After XRD (Rigaku Corporation RINT-TTR) analysis of the precipitates that had been produced from the over-saturation direction, selected precipitates were added into deionized water to approach solubility from the under-saturation direction.
The pH of the samples was adjusted between 6 and 9 using 10.0 mol dm−3 HCl (KANTO KAGAKU 18078-1B). Moderately reduced conditions were maintained using hydrazine monohydrate (Wako Pure Chemical Industries, Ltd. 081-00893). The ionic strength was 0.8 mol kg−1 for all nine samples. Solubility experiments were performed in 50 cm3 tempered hard-glass vials with screw cap (NICHIDEN-RIKA GLASS CO., Ltd SV-50M). The glass vials were sealed with polytetrafluoroethylene (NICHIAS Corporation TOMBO No. 9082) and placed into a constant temperature oven. The temperature was maintained at 348 K inside the oven.
The contents of the glass vial were stirred periodically until they were analyzed. After several different aging periods, ranging from 29 to 217 days, the glass vials were taken out of the oven, then kept at room temperature for 5 hours. The pH and Eh of the suspension were measured at 298 ± 1 K with a combination glass electrode (TOA, GST-5741C) that was calibrated against pH buffers and an oxidation–reduction potential electrode (TOA, PTS-5011C), respectively. Aliquots of the aqueous phase were withdrawn and filtered through ultrafilters with an effective 10,000 molecular-weight cut-off (ADVANTEC USY-1) at room temperature. Before collecting the filtrate for analysis, a small aliquot of the aqueous phase was passed through the filters to saturate any possible adsorption sites on the filters and filtration containers (this filtrate was discarded). Filtrates were acidified with 0.01 mol dm−3 nitric acid (HNO3) (KANTO KAGAKU 25831-08) and stored until they were analyzed for Se and Fe using ICP-MS with a detection limit of 1.27 × 10−9 mol dm−3 of Se and that of 1.79 × 10−9 mol dm−3 of Fe. The filtrate for the analysis to determine the Se concentration was oxidized by hydrogen peroxide (KANTO KAGAKU 18084-00) to prevent precipitation and volatilization of Se before the acidification with HNO3. The precipitate was separated by a 0.45 μm membrane filter, then dried at 348 K inside the oven until this was withdrawn for XRD analysis. The dried precipitate was taken out of the glovebox where the solubility experiments were carried out, then characterized by XRD analysis in the other glovebox.
2.2. Determination of pH
Ionic strength affects hydrogen ion activity (aH+). The observed pH (pHobs) measured with a combination glass electrode might be different from the correct pH corresponding to −log10 aH+ because of the difference between ionic strength of the calibration buffer and the samples used in this study. Therefore, instead of the pHobs values, the calculated values of −log10 aH+ were used to analyze the solubility data in this study. The −log10 aH+ value (pH) is calculated by
(5)
(5) where γH+ is the activity coefficient of H+ and [H+] is the concentration of H+.
The specific ion interaction theory (SIT) model [Citation10] gives a good estimation of the activity coefficient in an ionic medium of up to 3.5 mol kg−1 through the use of ion interaction coefficients (ϵ(i, j)). Because the concentration of Cl− was much higher than those of other anions in the samples, log10 γH+ is calculated using SIT as follows:
(6)
(6) where
(7)
(7)
D(T) is the Debye–Hückel term [Citation10], literature ϵ(H+, Cl−) value is 0.12 ± 0.01 kg mol−1 [Citation10], mCl- is the molality of Cl− and Im is the molal ionic strength. The values of A(T) calculated from the static dielectric constant and the density of water as functions of temperature and pressure are listed in TDB-2 [Citation10]. In the estimation of log10 γH+, the value of A(T) at a pressure of 1.00 bar for a temperature of 298 K was used to calculate the D(T) value because the pH measurements of the equilibrated suspensions were made at 298 ± 1 K.
Hydrogen ion concentrations for all samples were determined using the method developed by Rai and Yui [Citation11]. In this method, hydrogen ion concentrations are estimated with the correction factor “B” as follows:
(8)
(8)
The value of B needed to convert the pHobs reading to log10 [H+] was obtained by a titration procedure in which 10−pHobs are plotted against the moles of added free acid per liter. Because this correction factor varies with ionic strength but does not vary with hydrogen ion concentration at a fixed ionic strength, 0.80 mol kg−1 NaCl solution of the same ionic strength as the samples used in this study was titrated with 0.0100 mol dm−3 HCl solution. The raw experimental data are shown with open circles in . The solid line denotes the linear least-squares fit of data. The logarithm of the slope of this line corresponds to B given by
(9)
(9)
Figure 3. Titration of 0.80 mol kg−1 NaCl with 0.0100 mol dm−3 HCl using a combination glass electrode. [H+free, add] is the moles of added free acid per liter and [H+obs] = 10−pHobs, where pHobs is the observed pH measured with a combination glass electrode. [H+obs] as a function of [H+free, add] is shown as open circles. The solid line denotes the linear least-squares fit of data. The logarithm of the slope of this line corresponds to B = 0.03. This value is required to convert the pHobs reading to the −log10 [H+] value of the samples used in this study, using the following equation: −log10 [H+] = pHobs + B.
![Figure 3. Titration of 0.80 mol kg−1 NaCl with 0.0100 mol dm−3 HCl using a combination glass electrode. [H+free, add] is the moles of added free acid per liter and [H+obs] = 10−pHobs, where pHobs is the observed pH measured with a combination glass electrode. [H+obs] as a function of [H+free, add] is shown as open circles. The solid line denotes the linear least-squares fit of data. The logarithm of the slope of this line corresponds to B = 0.03. This value is required to convert the pHobs reading to the −log10 [H+] value of the samples used in this study, using the following equation: −log10 [H+] = pHobs + B.](/cms/asset/c0221a3a-c612-431e-9b6d-3a2e3c5f9033/tnst_a_1137245_f0003_b.gif)
As a result, the value of −log10 [H+] was determined by measuring pHobs and applying the correction factor determined above.
3. Results and discussion
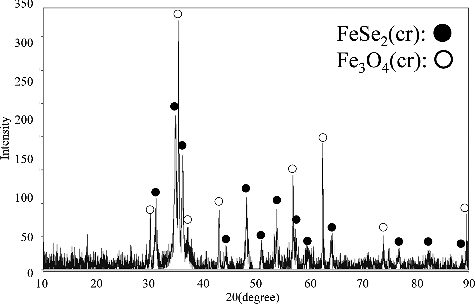
and show the XRD pattern of the solid sample from 158-day aged suspension produced from the over-saturation direction and that of the solid sample from 105-day aged suspension produced from the under-saturation direction, respectively. XRD analysis of the equilibrated precipitates showed the presence of FeSe2(cr) and magnetite (Fe3O4(cr)) and the absence of any other crystalline solids in all samples. There was a possibility of the precipitation of Fe(OH)2(cr). However, even if Fe(OH)2(cr) was formed, Fe(OH)2(cr) was considered to be oxidized to Fe3O4(cr) upon exposure to the atmosphere before XRD analysis because Fe(OH)2(cr) is readily oxidized by atmospheric oxygen. Iida et al. [Citation6] also reported the oxidation of Fe(OH)2(cr) soon after taking the equilibrated precipitates out of the glovebox.
Figure 4. XRD pattern of the solid sample from 158-day aged suspension produced from the over-saturation direction. The pH of this suspension was 6.08.
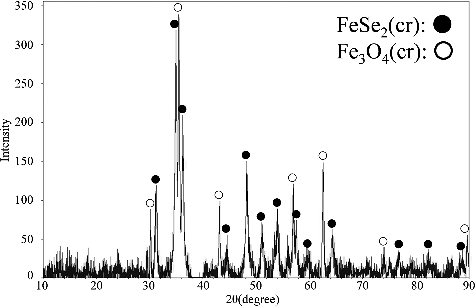
Figure 5. XRD pattern of the solid sample from 105-day aged suspension produced from the under-saturation direction. The pH of this suspension was 6.64.
shows a summary of the solubility experiments. The Eh and pH data shown in are plotted on the Eh–pH diagram of to identify the possible predominant Se species. As suggested in , the redox-pH conditions of all samples are in accordance with a stability field for Se42−. On the other hand, the Eh–pH diagram of , which is based on the thermodynamic database provided by Lemire et al. [Citation12], illustrates the stability ranges of the dominant Fe oxidation states and the predominant solution speciation in aqueous systems. Stability fields of Fe solids occupy the ranges of alkaline and neutral pH conditions in an Eh–pH diagram for Fe based on the above-mentioned thermodynamic database. In the preparation for the Eh–pH diagram of , the thermodynamic data of Fe solids were removed from the above-mentioned thermodynamic database, thereby making stability fields of Fe aqueous species visible. It can be seen from the Eh–pH diagram of that the redox-pH conditions of all samples fall in the region where Fe2+ is the dominant aqueous species. The dissolved Fe was likely to be present as the II oxidation state at the lower Eh of the samples than the Fe(II) to Fe(III) oxidation.
Table 1. Summary of solubility experiments of Se performed from both the over- and under-saturation directions in the presence of Fe under moderately reduced and neutral pH conditions.
Figure 6. Eh–pH diagram for Fe based on the thermodynamic data provided by Lemire et al. [Citation12]. The total concentration of Fe is 1×10−7 mol dm−3. The experimental data obtained from the over- and under-saturation directions are plotted as open circles and solid symbols, respectively.
![Figure 6. Eh–pH diagram for Fe based on the thermodynamic data provided by Lemire et al. [Citation12]. The total concentration of Fe is 1×10−7 mol dm−3. The experimental data obtained from the over- and under-saturation directions are plotted as open circles and solid symbols, respectively.](/cms/asset/33472333-e110-4eed-9350-da2c1d9fc8f4/tnst_a_1137245_f0006_b.gif)
The XRD patterns of the solid samples and the presence of Se42− and Fe2+ in the aqueous solutions, based on Eh–pH plots as discussed above, are not sufficient to prove that FeSe2(cr) is the equilibrating solid because there is a possibility that an amorphous solid or a finite amount of solid phase below the detection limit of XRD analyses is the solubility-controlling solid. However, if the concentrations of Se and Fe are controlled by the dissolution reaction in EquationEquation (3)(3)
(3) , then the log10K○2 values calculated from the experimental data of the different samples should be similar. Therefore, to test this hypothesis, the log10K○2 values for the reaction in EquationEquation (3)
(3)
(3) were calculated using EquationEquation (4)
(4)
(4) for all samples. The −log10 ae- value is calculated by
(10)
(10)
The activity of Se42− was determined from the Se concentration using the following equation:
(11)
(11)
The following equilibrium constant permits replacement of the activity of HSe− in EquationEquation (11)(11)
(11) by those of Se42−, H+ and e−:
(12)
(12)
The value of log10K○3 was calculated from ΔfG○m(Se42−) and ΔfG○m(HSe−). Iida et al. [Citation2] and Olin et al. [Citation13] reported ΔfG○m(Se42−) = 95.14 ± 0.17 kJ mol−1 and ΔfG○m(HSe−) = 43.471 ± 2.024 kJ mol−1, respectively. Combination with values for ΔfG○m(Se42−) and ΔfG○m(HSe−) provides the value of log10K○3 = −13.80 ± 1.42. As is the case for the Se42−/HSe− couple, the activity of Fe2+ was determined from the Fe concentration using the following equation:
(13)
(13)
The following equilibrium constant permits replacement of the activity of Fe(OH)+ in EquationEquation (13)(13)
(13) by those of Fe2+, H+ and H2O:
(14)
(14)
Lemire et al. [Citation12] reported the value of log10K○4 = − 9.100 ± 0.400. The values of log10 aH2O are calculated by
(15)
(15) where ϕ is the osmotic coefficient of the mixture and the summation extends over all solute species k with molality mk present in the solution [Citation13]. The following equations yield the osmotic coefficient for a mixed electrolyte [Citation14]:
(16)
(16)
(17)
(17)
(18)
(18)
(19)
(19) where fϕ is the Debye–Hückel term extended to include osmotic effects, z is the charge of ion and c is an index covering all cations, while a denotes all anions. The parameters of βca(0) and βca(1) define the second virial coefficient, and Ccaϕ defines the third virial coefficient [Citation15].
Using the SIT model, the activity coefficients of Se42−, HSe−, Fe2+ and Fe(OH)+ were calculated as follows:
(20)
(20)
(21)
(21)
(22)
(22)
(23)
(23)
The value of A(T) at a pressure of 1.00 bar for a temperature of 298 K [Citation10] was substituted in EquationEquation (7)(7)
(7) when calculating the above D(T) values. Available literature values for ϵ(Se42−, Na+), ϵ(HSe−, Na+) and ϵ(Fe2+, Cl−) are −0.03 ± 0.02 kg mol−1 [Citation2], −0.01 ± 0.10 kg mol−1 [Citation2] and 0.17 ± 0.01 kg mol−1 [Citation12], respectively. The value of ϵ(Fe(OH)+, Cl−) is not available in the literature. An average of all the ϵ(M+, Cl−) values reported in Lemire et al. [Citation12] was used as a substitution for ϵ(Fe(OH)+, Cl−), since the ion interaction coefficient is dependent on the charge of the ion [Citation10].
As can be found from , the log10K○2 values calculated from the experimental data of the different samples were all similar, independent of aging time. An average log10K○2 value of −17.07 was obtained. This finding suggests that (1) 29 days are long enough as aging time, (2) the dissolution reaction which controlled the concentrations of Se and Fe was essentially identical for all samples in spite of various redox-pH conditions and (3) equilibrium was established in all samples at the time of measuring the pH and Eh of the suspension at 298 ± 1 K, based on agreement between solubility data obtained from the over- and under-saturation directions. A slope analysis, which gives direct information on the solubility-controlling solid and allows the log10K○ value for the dissolution reaction to be determined, is therefore useful in the overall fit to the solubility data. The dissolution reaction of FenSe with Se42− and Fe2+ can be described as
(24)
(24)
The K○5 value of this dissolution reaction is given by
(25)
(25)
The following equations are obtained by converting EquationEquation (25)(25)
(25) :
(26)
(26) where
(27)
(27)
(28)
(28)
From EquationEquation (26)(26)
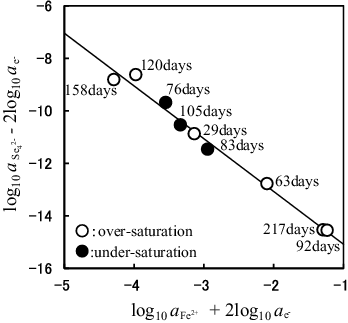
(26) , one would expect a graph of Y vs. X to be linear with an intercept of log10K○5 and a slope of (−4n), allowing n for the reaction and log10K○5 to be determined. A plot of the Y values vs. the X values is shown in , where the solid line represents the linear least-squares fit of data. The slope of this line corresponds to −4n given by
(29)
(29)
Figure 7. Determination of the n value for FenSe which is the solubility-controlling solid and the log10K○5 value for FenSe dissolution reaction represented by (4FenSe(cr) ⇌ 4nFe2+ + Se42− + (8n − 2)e−) using the SIT model. The value of () is plotted against the value of (
). Solubility data obtained from the over- and under-saturation directions are shown as open circles and solid symbols, respectively. The solid line denotes the linear least-squares fit of data. The slope and intercept of this line correspond to −4n = −2.01 ± 0.09 and log10K○5 = −17.09 ± 0.28, respectively.
Substituting the above value for n in EquationEquation (24)(24)
(24) yields the dissolution reaction in EquationEquation (3)
(3)
(3) , indicating that (1) log10K○5 is essentially identical to log10K○2, and (2) FeSe2(cr) was most likely the solubility-controlling solid. The intercept of the solid line in corresponds to log10K○5 given by
(30)
(30)
The above log10K○2 value is applicable to a temperature of 298 K. If equilibrium was assumed to be established at 348 K before the samples were taken out of the oven, then it can be seen that the equilibrium was reached again rather rapidly (< 5 hours) at 298 ± 1 K. Apparently, the FeSe2(cr) solubility at 298 K is not significantly different from that at 348 K.
As can be seen from , significant differences were found for the Fe concentration between the different samples, although the Se concentrations were not significantly different. As can be implied from EquationEquation (4)(4)
(4) , only Eh is predicted to affect concentrations of Se and/or Fe. Different Fe concentrations are considered to be responsible for differences in Eh. It seems that the influence of Eh on the concentration of Fe was greater than that on the concentration of Se.
An attempt was also made to see whether the Fe3O4(cr) that was detected by XRD analysis controlled the solubility of Fe. When Fe3O4(cr) controlled concentrations of Fe2+, the following equilibrium constant permits predictions of the activity of Fe2+:
(31)
(31)
The value of log10K○6 was calculated from ΔfG○m(Fe2+), ΔfG○m(H2O) and ΔfG○m(Fe3O4(cr)). Lemire et al. [Citation12] reported ΔfG○m(Fe2+) = −90.719 ± 0.641 kJ mol−1, ΔfG○m(H2O) = −237.140 ± 0.041 kJ mol−1 and ΔfG○m(Fe3O4(cr)) = −1012.719 ± 1.609 kJ mol−1. Combination with values for ΔfG○m(Fe2+), ΔfG○m(H2O) and ΔfG○m(Fe3O4(cr)) provides the value of log10K○6 = 36.439 ± 0.440. The activity of Fe2+ in the samples was found not identical to that predicted by the above log10K○6 value thereby indicating that Fe3O4(cr) did not control the solubility of Fe in any samples. On the other hand, the Eh–pH diagram of , which is based on the thermodynamic database provided by Lemire et al. [Citation12], illustrates the stability ranges of the dominant Fe oxidation states and the predominant solution speciation in aqueous systems. The Eh and pH data shown in are plotted on the Eh–pH diagram of . As suggested in , the redox-pH conditions of all samples are not in accordance with a stability field for Fe3O4(cr), indicating that Fe3O4(cr) was not thermodynamically stable in all samples. No evidence that Fe3O4(cr) controlled the solubility of Fe was observed.
Figure 8. Eh–pH diagram for Fe based on the thermodynamic data provided by Lemire et al. [Citation12]. The total concentrations of Fe are 1 × 10−7 mol dm−3 (solid line) and 1 × 10−4 mol dm−3 (dotted line). The experimental data obtained from the over- and under-saturation directions are plotted as open circles and solid symbols, respectively.
![Figure 8. Eh–pH diagram for Fe based on the thermodynamic data provided by Lemire et al. [Citation12]. The total concentrations of Fe are 1 × 10−7 mol dm−3 (solid line) and 1 × 10−4 mol dm−3 (dotted line). The experimental data obtained from the over- and under-saturation directions are plotted as open circles and solid symbols, respectively.](/cms/asset/af3809eb-8270-403e-94a9-87e5a5e2405d/tnst_a_1137245_f0008_b.gif)
The log10K○2 value determined above was compared with that calculated from the available thermodynamic data. The log10K○2 value which is valid at T0 can be calculated by
(32)
(32) When calculating the log10K○2 value, ΔfG○m(Fe2+) = −90.719 ± 0.641 kJ mol−1 reported in Lemire et al. [Citation12] and ΔfG○m(Se42−) = 95.14 ± 0.17 kJ mol−1 reported in Iida et al. [Citation2] were used. The value of ΔfG○m(FeSe2(cr)) was calculated using EquationEquation (2)
(2)
(2) . Olin et al. [Citation13] reported ΔfH○m(FeSe2(cr)) = −108.700 ± 15.000 kJ mol−1, S○m(FeSe2(cr)) = 86.800 ± 1.000 J K−1 mol−1 and S○m(Se(cr)) = 42.090 ± 0.330 J K−1 mol−1. The value of S○m(Fe(cr)) = 27.085 ± 0.160 J K−1 mol−1 reported in Lemire et al. [Citation12] in combination with these values for ΔfH○m(FeSe2(cr)), S○m(FeSe2(cr)) and S○m(Se(cr)) provides the value of ΔfG○m(FeSe2(cr)) = −101.406 ± 15.004 kJ mol−1. Using EquationEquation (32)
(32)
(32) , log10K○2 was calculated to be −20.41 ± 5.26. The average value thus calculated from the thermodynamic data is approximately three orders of magnitude lower than the log10K○2 value (−17.09 ± 0.28) determined in this study. However, our value falls within the uncertainty limits of the calculated value. The large uncertainty in the calculated log10K○2 value arises primarily from the uncertainty in ΔfH○m(FeSe2(cr)). Our value is much more precise with a smaller degree of uncertainty. In the past, the large uncertainty in the FeSe2(cr) solubility caused the complications in evaluating Se concentrations in repository environments. However, the log10K○2 value determined in this study is expected to remove such complications.
4. Conclusions
Predictions based on the available thermodynamic data suggest that FeSe2(cr) is expected to control Se concentrations under reducing repository environments. Despite the importance of the FeSe2(cr) solubility in the safety assessment of the geological disposal system, no studies wherein its solubility was measured are reported. Complication in evaluating Se concentrations in repository environments arose from the large uncertainty in the calculated K○1 value for FeSe2(cr) dissolution reaction as well as a lack of direct evidence of a solubility limit by FeSe2(cr) under reducing conditions.
Previous studies failed in forming FeSe2(cr) or measuring the concentrations of Se or Fe which are sensitive to variations in Eh and/or pH. In this study, aging at 348 K was used to promote the formation of FeSe2(cr) and moderately reduced and neutral pH conditions to produce the precipitation of Se42− and Fe2+ as FeSe2(cr) were maintained to measure the Se and Fe concentrations controlled by the following dissolution reaction of FeSe2(cr):
Nine different sets of experiments were carried out which ranged in aging periods from as low as 29 days to as long as 217 days and varied in approach to equilibrium (under- or over-saturation directions). XRD analysis detected only FeSe2(cr) as the Se solid phase in the equilibrated precipitates. Because the solubility data were independent of aging time, 29 days were considered to be long enough as aging time to form FeSe2(cr). Agreement between solubility data obtained from the over- and under-saturation directions suggests that equilibrium was established at 298 ± 1 K at the time of analyzing the samples. The thermodynamic interpretations using the SIT model showed that Eh values and the concentrations of Se and Fe are well represented by the above reaction, providing the value of log10K○2 = −17.09 ± 0.28 for its reaction, which is applicable to a temperature of 298 K and falls within the uncertainty limits of the log10K○2 value calculated from the available thermodynamic data. The large uncertainty in the calculated log10K○1 and log10K○2 values arises primarily from the uncertainty in ΔfH○m(FeSe2(cr)). Our value is much more precise with a smaller degree of uncertainty. The data reported in this study are expected to remove the complications mentioned above.
| Nomenclature | ||
| ai | = | activity of ion i |
| γi | = | activity coefficient of ion i |
| zi | = | charge of ion i |
| mi | = | molality of ion i (amount of ion i divided by the mass of the solvent, mol kg−1) |
| [i] | = | concentration of ion i (amount of ion i divided by the volume of the solution, mol dm−3) |
| R | = | gas constant |
| F | = | Faraday constant |
| T | = | absolute temperature (K) |
| T0 | = | reference temperature ( = 298.15 K) |
| Im | = | ionic strength of solution ( |
| D(T) | = | Debye–Hückel term |
| A(T) | = | Debye–Hückel parameter (values taken from TDB-2 [Citation10]) |
| ϵ(i, j) | = | ion interaction coefficient between ion i and ion j of opposite charge (kg mol−1) |
| Eh | = | oxidation–reduction potential against the standard hydrogen electrode (V vs. SHE) |
| pH | = | negative logarithm of hydrogen ion activity(= −log10 aH+) |
| pHobs | = | the pH value measured with a combination glass electrode |
| ΔfG○m | = | the standard molar Gibbs energy of formation from the elements in their reference state (kJ mol−1) |
| ΔfH○m | = | the standard molar enthalpy of formation from the elements in their reference state (kJ mol−1) |
| S○m | = | the standard molar entropy (J K−1 mol−1) |
| K○ | = | the equilibrium constant of the reaction |
Acknowledgments
The authors wish to thank Dr Rai (Rai Enviro-Chem, LLC), Dr Kobayashi (Kyoto University), Dr Kitamura (Japan Atomic Energy Agency), Dr Iida (Japan Atomic Energy Agency), Dr Fujiwara (Japan Atomic Energy Agency) and Dr Yoshida (NESI Co., Ltd.) for the valuable discussions and suggestions to prepare this manuscript, Mr Hayashi (Inspection Development Co. Ltd.) and Ms Hoshino (Inspection Development Co. Ltd.) for ICP-MS analysis and Mr Ohuchi (Inspection Development Co. Ltd.) and Mr Takahashi (Inspection Development Co. Ltd.) for XRD analysis.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Kitamura A, Doi R, Yoshida Y. Update of JAEA-TDB: update of thermodynamic data for palladium and tin, refinement of thermodynamic data for protactinium, and preparation of PHREEQC database for use of the Brønsted-Guggenheim-Scatchard model. Tokai-mura: Japan Atomic Energy Agency; 2014, (Report no. JAEA-Data/Code 2014-009).
- Iida Y, Yamaguchi T, Tanaka T, et al. Solubility of selenium at high ionic strength under anoxic conditions. J Nucl Sci Technol. 2010;47:431–438.
- Doi R, Tachikawa H, Yui M. Transformation of selenium solid phase in the presence of iron under reducing conditions. J Nucl Sci Technol. 2010;47:278–285.
- Kitamura A, Shibata M, Kitao H. Solubility measurement of iron-selenium compounds under reducing conditions. In: Oversby VM, Werme LO, editors. Proceedings of Material Research Society Symposium; 2004 Jun 15–19; Kalmarsalen (Sweden): Materials Research Society.
- Shibutani S, Yoshikawa H, Yui M. Solubility measurement of Se in Se-H2O system under reducing condition. Tokai-mura: Power Reactor and Nuclear Fuel Development Corporation; 1995, (Report no. PNC TN8410 94-204).
- Iida Y, Yamaguchi T, Tanaka T, et al. Determination of the solubility limiting solid of selenium in the presence of iron under anoxic conditions. Proceedings of International Workshop on Mobile Fission and Activation Products in Nuclear Waste Disposal; 2007 Jan 16–19; Hôtel Barrière L’Hermitage, La Baule (France): OECD Publications; 2009.
- Doi R, Kitamura A, Yui M. JAEA thermodynamic database for performance assessment of geological disposal of high-level and TRU wastes: selection of thermodynamic data of selenium. Tokai-mura: Japan Atomic Energy Agency; 2009, ( Report no. JAEA-Review 2009-051).
- Warren CG. The synthesis of ferroselite from an aqueous solution at low temperature. Econ Geol. 1968;63:418–419.
- Yui M, Sasamoto H, Arthur RC. Groundwater evolution modeling for the second progress performance assessment (PA) report. Tokai-mura: Japan Nuclear Cycle Development Institute; 1999, (Report no. JNC TN8400 99-030).
- Grenthe I, Mompean F, Spahiu K, et al. TDB-2 guidelines for the extrapolation to zero ionic strength. Issy-les-Moulineaux (France): OECD Nuclear Energy Agency; 2013.
- Rai D, Yui M. Thermodynamic data development using the solubility method (joint research). Tokai-mura: Japan Atomic Energy Agency; 2013, (Report no. JAEA-Technology 2013-002).
- Lemire RJ, Berner U, Musikas C, et al. Chemical thermodynamics of iron part 1. Paris: OECD Publications; 2013.
- Olin Å, Noläng B, Öhman LO, et al. Chemical thermodynamics of selenium. Amsterdam: Elsevier; 2005.
- Pitzer KS, Kim JJ. Thermodynamics of electrolytes. IV. Activity and osmotic coefficients for mixed electrolytes. J Am Chem Soc. 1974;96:5701–5707.
- Pitzer KS, Mayorga G. Thermodynamics of electrolytes. II. Activity and osmotic coefficients for strong electrolytes with one or both ions univalent. J Phys Chem. 1976; 77:2300–2308.