
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Ion exchange is one of the most useful separation methods. Certain ion exchange resins have been developed for use in reprocessing technology. The purpose of this study is to evaluate the U selectivity of a benzimidazole-type anion exchange resin and its potential for the separation of U from a solution of simulant fuel debris in HCl. The selectivity of the benzimidazole-type anion exchange resin for adsorption of the major elements present in fuel debris in HCl solution (such as in debris from the Fukushima Daiichi nuclear power plant) was analyzed, and the tendencies were discussed from the viewpoint of complexation. Moreover, chromatographic separation of U from the other elements was evaluated using the resin. The results indicate that this type of resin has high U separation selectivity. Consequently, the authors developed a novel strategy involving the use of this resin for U separation in conjunction with chlorination of hardly soluble materials by using atmospheric-pressure nonthermal plasma. This strategy is also proposed to be applicable for stabilizing waste forms and for nuclear fuel material accounting, such as for debris from the Fukushima nuclear power plant and other complex hardly soluble nuclear materials.
1. Introduction
Methods for conditioning fuel debris generated by the accident at the Fukushima Daiichi nuclear power plant, hereafter referred to as 1F, and other accident products, such as TMI-2 debris and Chernobyl fuel containing masses, have been discussed. However, an effective conditioning method has not yet been developed [Citation1]. Although aqueous reprocessing is one of the most reliable methods for conditioning fuel debris, the application of this method to fuel debris treatment is hampered by two major technical limitations. First, dissolving the fuel debris is difficult; e.g. (U,Zr)O2 solid solution, the main component of TMI-2 debris, is a hardly soluble material [Citation2,Citation3]. Although no fuel debris has yet been confirmed in 1F, the generation of (U,Zr)O2 solid solution is expected [Citation4]. The second problem concerns the separation of nuclear fuel materials. Fuel debris contains large amounts of structural materials such as stainless steels, zircaloy, and concrete. Although the solvent extraction-based Plutonium, Uranium, Reduction, Extraction (PUREX) process is an established and commercially used technology, it requires strict solution adjustment and is geared towards large-scale processing. Smaller scale processing systems, e.g. with a capacity of 20 tHM/y, are more appropriate for treating 1F debris than large-scale (e.g. 800 tHM/y) commercial reprocessing plants because the amount of debris is expected to be about 200 tHM [Citation5].
The present authors have focused on developing a chemical transformation method for converting hardly soluble materials into easily soluble ones, such as chlorides, by using atmospheric-pressure nonthermal plasma (APNTP) [Citation6]. The principle has been verified using nonradioactive materials and its application to fuel debris has been evaluated. The authors proposed that chemically transformed fuel debris can be dissolved in water or HCl solution. Ion exchange can be used for the separation of fuel debris solutions because it is capable of the selective separation of specific elements from solutions containing various elements and is appropriate for small-scale processing plants. From the viewpoint of fuel debris conditioning, quantitative determination of nuclear fuel materials such as U and Pu is important for both accounting and stabilization of waste materials.
Application of anion exchange resins such as tertiary pyridine-type anion exchange resins to the advanced Optimization by Recycling Instructive Elements (ORIENT) cycle, which is an innovative reprocessing concept for the recovery of U, minor actinides, and Pt group metals (PGM), has been discussed, because these types of resins can selectively separate the aforementioned species [Citation7,Citation8]. Moreover, the resins are embedded in porous silica beads and have high radiation resistance [Citation7]. A benzimidazole-type anion exchange resin embedded in porous silica beads (fabricated by Asahi Kasei), termed AR-01, also has high radiation resistance and has been evaluated for uranium enrichment and separation of U from a few fission products (FPs) in HCl solution [Citation9,Citation10]. It was verified in the development of uranium enrichment that AR-01 has resistance to high temperature and redox reaction [Citation11]. Because the absorbability of ions for ion exchangers drastically depends on their valence, AR-01 might be useful for separation with adjustment of redox potential. From the above, this resin is considered a promising medium for the separation of U and Pu from a fuel debris solution containing FPs, constructional materials, and transition metals. In this study, the efficacy of AR-01 for the adsorption and separation of U from a simulant fuel debris solution is studied. Furthermore, based on the experimental results and a previous study on the chlorination of hardly soluble materials with APNTP [Citation6], a process for the treatment of fuel debris is proposed.
2. Experimental detail
2.1. Evaluation of distribution coefficient and ultraviolet–visible absorption analysis
The distribution coefficient of certain elements that are expected in 1F debris on AR-01 in HCl (EquationEquation (1(1)
(1) )) was measured. The concentrations of HCl used herein were 1, 3, 6, and 9 M. AR-01 was obtained from Asahi Kasei and possessed N-methylbenzimidazole and N,N′-dimethylbenzimidazolium as the exchange sites. The structure and properties of AR-01 have been presented elsewhere [Citation8,Citation12], but some properties that were measured in this study are summarized in . The metal ions assayed include U(VI) as the actinide element and Zr(IV), Sn(IV), Fe(III), Cr(III), Ni(II), and Co(II) as the main constructional elements. The main FPs present in the 1F fuel were estimated by ORIGEN2 [Citation13]. The FPs used in this test include Sr(II), Cs(I), and Ba(II) as alkali or alkaline earth elements; La(III), Ce(III), Pr(III), Nd(III), Sm(III), and Gd(III) as lanthanides; Ru(III) and Pd(II) as the PGMs; and Mo(VI) as the transition metal. All metallic ions except Ru(III) and U(VI) were prepared by dissolving their nonradioactive solid-state chlorides or oxychlorides in HCl; Ru(III) and U(VI) were prepared from RuCl3 in HCl and 10 ppm U-containing HNO3 solution, respectively.
Table 1. Properties of AR-01.
For the batch adsorption experiments, 0.5 g of AR-01 was added to a vial of a 10 mL HCl solution containing 2 mM of either a single metallic ion or a mixture of metallic ions with no mutual redox reactions. The solutions were maintained at 25 °C in a water bath for 2 h and shaken mechanically. AR-01 was then removed using a syringe filter with a pore size of 0.45 µm. The concentration of metal ions in the solution before and after adsorption was measured using inductively coupled plasma mass spectrometry (ICP-MS, Agilent Technology, 7700). The distribution coefficient (Kd) was calculated from the measurement results as follows:
(1)
(1) where C0 and C indicate the concentration of the metallic ions in HCl before and after adsorption, respectively, and V and m indicate the volume of HCl and the volume of the resin, respectively.
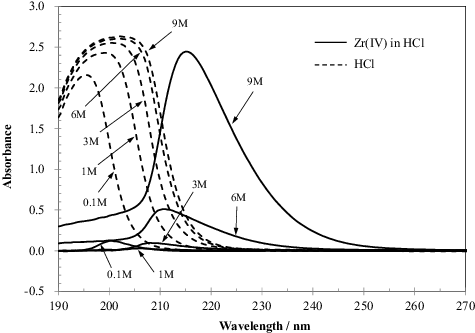
Zr(IV) in HCl was analyzed by ultraviolet–visible (UV–vis) spectroscopy (Shimadzu, UV-2450) to evaluate the transformation of the Zr(IV) complexes and the relationship between the absorption spectra and Kd values. The Zr(IV) solution was prepared using ZrCl4 and was used for analysis within a few hours of preparation. The concentrations of HCl were 0.1, 1, 3, 6, and 9 M, and the concentration of Zr in the HCl solution was 4.3 mM. A deuterium lamp was used as the UV light source and a tungsten-halogen lamp was used as the visible light source for UV–vis spectroscopy. The test solution was placed in a square quartz cell having an optical length of 10 mm; two cells one each for the reference sample were used in the device. The UV–vis absorption of HCl at each concentration was measured with pure water as the reference and HCl as the target analyte. The absorption of Zr in HCl was measured against an HCl solution of the same concentration as the blank.
2.2. Chromatography experiment
The separation condition for the chromatography experiment was decided by calculating the separation factor α from the distribution coefficients as follows:
(2)
(2) where Kd,U is the distribution coefficient of U, the subscript M indicates a metallic ion, and Kd,M is the distribution coefficient of the metallic ion.
AR-01 was charged into a glass column with height 290 mm and diameter 8 mm. Before the chromatographic process, 200 cm3 of 9, 0.5, and 6 M HCl was passed through the column to remove any adsorbed metallic ions from the resin and exchange the adsorbed anions with the functional group Cl−. The chromatographic experiments were performed with a simulant high-level liquid waste containing the relevant elements at room temperature. First, 6 cm3 of the feed solution was introduced into the column using a high-pressure pump at a flow rate of 1.0 cm/min (0.5 cm3/min). Subsequently, 6 and 0.5 M HCl solutions (200 cm3) were sequentially introduced into the column at a rate of 2.0 cm/min (1.0 cm3/min). The reasons for selecting these HCl concentrations for U(VI) adsorption or elution are discussed in Section 3.2. The flow rate of the feed solution is slower than that of the 6 and 0.5 M HCl solutions because, in the case of low feed volume, ions are uniformly adsorbed in the upper region of the column and a smooth chromatogram is obtained. The outflow liquid (in aliquots of 2 or 4 cm3) was collected using a fraction collector, and the concentrations of the metal ions in the samples were measured by ICP-MS.
3. Results and discussion
3.1. Distribution coefficients and ultraviolet–visible absorption spectra
The distribution coefficient plots are shown in . Alkali, alkaline earth, and lanthanide elements show almost no adsorption on the resin, because these ions remain in the cationic state without forming chloro complexes. The values of the distribution coefficients for all elements, except elements that are not adsorbed or elements that form distinct complexes, depend on the following three reactions: complex formation (Equation3(3)
(3) ), anion exchange (Equation4
(4)
(4) ), and proton association (Equation5
(5)
(5) ) [Citation14]:
(3)
(3)
(4)
(4)
(5)
(5)
where M and R represent the metal and resin type, respectively. The distribution coefficients of U(VI), Fe(III), and Co(II) show the same tendency, wherein Kd increases with HCl concentration. The aforementioned ions form chloro complexes such as UO2Clx2−x, FeCl4−, and CoCl42−. Their complex formation and anion exchange processes are based on reactions (Equation3
(3)
(3) ) and (Equation4
(4)
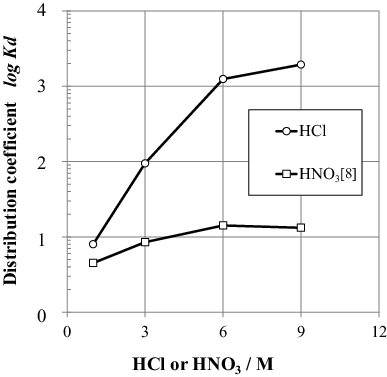
(4) ). The concentrations of chloro complexes increase with increasing Cl− concentration, thus facilitating the anion exchange reactions. The distribution coefficients of U(VI) in HCl and HNO3, measured by Wei et al. [Citation8], are compared in . The Kd values increase with concentration in both acids. Uranium chloride complexes, UO2Clx2−x, are more strongly adsorbed on AR-01 than the uranium nitride complex UO2(NO3)3− [Citation15]. This result shows that HCl may be useful for the separation of U(VI) from FPs and other elements in the spent fuel solution because of the high Kd value of U(VI) and the selectivity of the ion exchange resin for U(VI).
Figure 1. Distribution coefficients of the main elements in 1F debris with AR-01 in HCl.
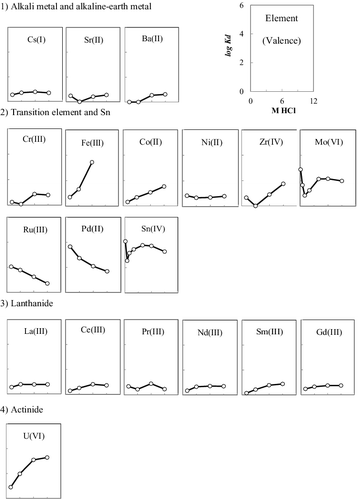
Figure 2. Distribution coefficients of U(VI) for interaction with AR-01 in HCl and HNO3.
The Kd value trends of Mo(VI), Zr(IV), and Sn(IV) are similar to that of U(VI) for acid concentrations above 0.5–3 M, as shown in . The distribution coefficient of Mo(VI) is minimum at 1 M and increases with decreasing HCl concentration. The major complex type of Mo(VI) in HCl changes from [H3MoO4]+ to MoO2Cl2 at an HCl concentration of ∼3 M, and MoO2Cl3− predominates at HCl concentrations above 3 M, as shown in (a) [Citation16]. The anion exchange reaction is dominated by MoO2Cl3−. The major type of Sn(IV) complex for HCl concentrations above 1 M is SnCl62−, and the fraction of this species increases with increasing HCl concentration, as shown in (b) [Citation17].
Figure 3. Distribution diagrams for Mo(VI) and Sn(IV) as a function of [Cl−] calculated from stability constants. (a) Distribution diagram for Mo(VI) as a function of [Cl−] at 298 K. The stability constants between Mo(VI) species and H+ or Cl− are β1 = 104.21, β2 = 108.21, and β3 = 109.14 and β[Mo(OH)5H2O+ + Cl− ⇄ Mo(OH)5Cl + H2O] = 10−0.89, β[Mo(OH)5Cl + H+ + Cl− ⇄ MoO2Cl2 + 3H2O] = 10−1.42, and β[MoO2Cl2 + Cl− ⇄ MoO2Cl3−] = 10−2.64, respectively [Citation15]. (b) Distribution diagram for Sn(IV) as a function of [Cl−]. Temperature is unknown. The stability constants between Sn(IV) species and Cl− are β1 = 103.59, β2 = 106.24, β3 = 108.44, β4 = 109.14, β5 = 1010.62, and β6 = 1011.75 [Citation16].
![Figure 3. Distribution diagrams for Mo(VI) and Sn(IV) as a function of [Cl−] calculated from stability constants. (a) Distribution diagram for Mo(VI) as a function of [Cl−] at 298 K. The stability constants between Mo(VI) species and H+ or Cl− are β1 = 104.21, β2 = 108.21, and β3 = 109.14 and β[Mo(OH)5H2O+ + Cl− ⇄ Mo(OH)5Cl + H2O] = 10−0.89, β[Mo(OH)5Cl + H+ + Cl− ⇄ MoO2Cl2 + 3H2O] = 10−1.42, and β[MoO2Cl2 + Cl− ⇄ MoO2Cl3−] = 10−2.64, respectively [Citation15]. (b) Distribution diagram for Sn(IV) as a function of [Cl−]. Temperature is unknown. The stability constants between Sn(IV) species and Cl− are β1 = 103.59, β2 = 106.24, β3 = 108.44, β4 = 109.14, β5 = 1010.62, and β6 = 1011.75 [Citation16].](/cms/asset/233e34a9-b862-4975-8abf-5b52c2514978/tnst_a_1150219_f0003_b.gif)
In solution, Zr adopts various structures and the complexation of Zr with chloride ions is a topic that is still under discussion. The stability constants for Zr(IV) in a mixture of HClO and HCl were previously measured, indicating the presence of four complexes ZrClx4−x (x = 1 to 4) [Citation18]. This complexation was not consistent with the Kd value trend observed in this study because these cationic complexes do not react with the anion exchange resins. UV–vis absorption spectra of HCl and Zr(IV) in HCl were measured to evaluate the complex speciation of Zr(IV) in HCl and its relation with the Kd values determined in this study. The spectra of only HCl and Zr(IV) in HCl are shown in . Both spectra show a peak in the UV region, and the location of the absorption maximum differs with HCl concentration. The Zr(IV) spectra can be divided into three types based on the absorption maxima: at an HCl concentration of 0.1 M, a peak is observed around 200 nm; in 1 M HCl, a low-intensity peak is observed at a slightly longer wavelength; for HCl concentrations of 3, 6, and 9 M, a peak occurs between 208 and 215 nm and the peak wavelength increases with increasing HCl concentration. These differences indicate that there are at least two kinds of complexes and that the complex type changes around 1 M HCl. This tendency roughly corresponds to that of the Kd values. The complex speciation of Zr(IV) in HCl was also evaluated by extended X-ray absorption fine structure (EXAFS) analysis [Citation19,Citation20]. The complexation of Zr in HCl at HCl concentrations above 0.03 M could be divided into three phases: in the first phase, {[Zr4(OH)x(20−x)H2O](16−x)+}n is transformed into [Zr4(OH)816H2O]8+ between 0.03 and 0.5 M HCl; in the second phase, [Zr4(OH)816H2O]8+ exists stably between 0.5 and 1.0 M HCl; in the third phase, [Zr3(OH)x](12−x)+ (x = 4 or 5) exists stably at concentrations above 1.0 M HCl. In these experiments, the Zr concentration was above 0.1 M when the HCl concentration was above 1 M, and the formation of the Zr monomer in dilute Zr and concentrated HCl was suggested. Although the suggested complexation of Zr corresponds to the UV–vis spectral data, the suggested complexes are cations, which cannot react with the anion exchange resins. Therefore, the following complexation is proposed: [Zr4(OH)816H2O]Clx(x−8)− transforms to [Zr3(OH)y]Clz(y+z −12)− (y = 4 or 5) around 1 M HCl, wherein the value of z increases with HCl concentration.
Figure 4. UV–vis absorption spectra of HCl and Zr(IV) in HCl. The Zr(IV) concentrations are 4.3 mM in the spectrum measurements of Zr(IV) in HCl; the HCl concentrations are 0.1, 1, 3, 6, and 9 M.
The distribution coefficients of the PGMs are also shown in . The Kd of Pd(II) is more than 1000 in 1 M HCl, but decreases with increasing HCl concentration, and finally becomes less than 100 in 9 M HCl. Strong complexation between Pd(II) and the benzimidazole-type anion exchange resin AR-01 has already been reported [Citation21]. PdCl42− and PdCl3− are the main Pd(II) species when the HCl concentration is above 1 M [Citation22]. The complex formation reactions of the Pd(II) ion and the ion exchange reactions with the benzimidazole-type anion exchange resin can be represented as follows:
(6)
(6)
(7)
(7)
The Kd value of Ru(III) decreases with increasing HCl concentration because of the equilibrium between the formation of the metal ion complex and the adsorption of Cl− onto the resin. Rh(III) is also a PGM and its Kd value is expected to be low [Citation21]. This is confirmed from the results of the chromatography experiment.
Similar to U, separation and measurement of the mass inventory of Pu are also necessary for material control and nuclear fuel materials accounting. The distribution behavior of each element on the benzimidazole-type anion exchange resin used in this study is similar to that observed with the tertiary pyridine resin [Citation14]. The distribution coefficient of Pu(IV) for interaction with the tertiary pyridine resin is almost zero for HCl concentrations below 3 M; this value increases with increasing HCl concentration and reaches 100 in 9 M HCl. This tendency is similar to that observed for U(VI) and the elution behavior of Pu(IV) seems similar to that of U(VI). Therefore, the Kd value of Pu(IV) for interaction with the benzimidazole-type anion exchange resin would be similar to that for its interaction with the tertiary pyridine resin, and the separation behavior of Pu(IV) would be similar to that of U(VI) from FPs and structural materials.
3.2. Chromatography
The chromatographic conditions for separating U(VI) from other materials depend on the Kd and α values. Because the Kd value of U(VI) in 6 M HCl is more than 1000 and the α values of Cr(III), Co(II), Ni(II), Sr(II), Zr(IV), Cs(I), Ba(II), and the lanthanides are more than 100, these ions will be separated from U(VI) by chromatography using 6 M HCl. It is difficult to use high HCl concentrations, such as 9 M, in actual plants because of corrosion. Therefore, 6 M HCl was selected for separation of U(VI) from other materials by adsorption onto the resin. After the adsorption of U(VI) onto the resin, the adsorbed U(VI) will be eluted with 1 M HCl; however, it will be difficult to separate Mo(VI) and Fe(III) from U(VI) because the α values of Mo and Fe are 1.2 and 1.6, respectively. Therefore, 0.5 M HCl was selected for U(VI) elution and Mo(VI) separation because the Kd value of Mo(VI) is relatively high in 0.5 M HCl.
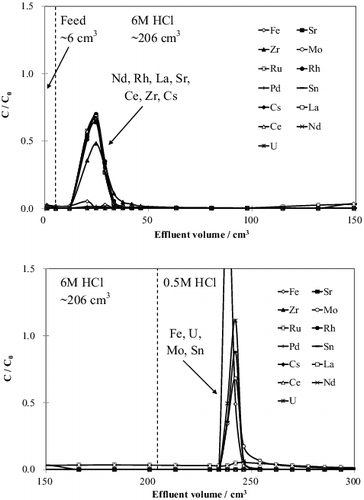
The chromatogram obtained in this test is shown in . After the inflow of 6 M HCl, Nd(III), Rh(III), La(III), Sr(II), Ce(III), Zr(IV), and Cs(I), which have low Kd values in 6 M HCl, were eluted simultaneously. Large amounts of Zr from cladding tubes is expected to be present in fuel debris in conjunction with U; thus, the possibility of separating Zr(IV) from U(VI) was verified. Furthermore, the low Kd value of Rh(III) in 6 M HCl and the separation from U were shown. Small amounts of Fe ion were also detected with the aforementioned elements. Fe(II) or Fe(III) may have been adsorbed onto the resin before the chromatographic experiment because the eluted amount of Fe ions was greater than that discharged into the column. After the inflow of 0.5 M HCl, Fe(III), U(VI), Sn(IV), and Mo(VI) were eluted. Although the separation of Mo(VI) and Sn(IV) from U(VI) was expected in 0.5 M HCl, Mo(VI) and Sn(IV) were both eluted at roughly the same time. If necessary, further study should be performed to investigate whether U can be separated from Fe, Sn, and Mo. Continuous elution of Pd(II) during the column experiment was confirmed, and it was found that more than 90% of Pd(II) remained on the column after the experiment. Because Pd(II) forms strong complexes with the benzimidazole-type resin, as mentioned above, the rate of formation and dissociation of the complexes between Pd(II) and the benzimidazole-type resin, as shown in reaction (Equation7(7)
(7) ), is slower than that in the normal ion exchange reaction (Equation4
(4)
(4) ). Therefore, in order to prevent the accumulation of Pd(II) in the column, allow reuse of the column for the recovery of U(VI), and prevent the destabilization of vitrified radioactive waste generated by melting AR-01 with adsorbed U(VI) [Citation23], specific separation of Pd(II) must be carried out before reaching this condition. The results confirmed that Ru(III) is also continuously eluted, similar to Pd(II), and that approximately 16% of the Ru(III) remained on the column after the experiment. This result shows that both Ru(III) and Pd(II) plausibly form strong complexes with the benzimidazole-type resin. The main complex species of Ru(III) is RuCl63−, and the association between RuCl63− and the benzimidazole-type resin has been described elsewhere [Citation21]. Thus, pre-separation of Ru(III) and Pd(II) from the AR-01 column is required prior to U separation.
Figure 5. Chromatogram for the simulant fuel debris solution.
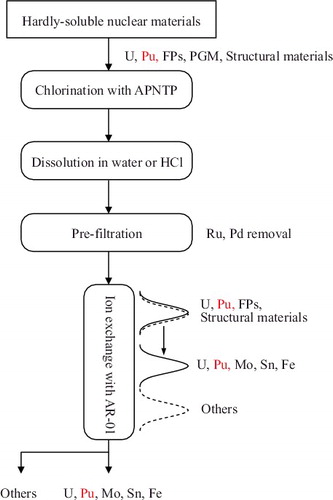
Based on the results of this work and previous studies [Citation6], we propose an appropriate process for the separation of U(VI) and Pu(IV) from hardly soluble nuclear materials using AR-01, as shown in . In step I, hardly soluble nuclear materials are chemically transformed into chlorides by APNTP. Subsequently, the transformed materials are dissolved in either water or HCl, and the PGMs, especially Pd and Ru, are separated. Tertiary pyridine-type resins are candidates for separating PGMs from U(VI) and other elements. S. Koyama et al., who evaluated the separation performance of tertiary pyridine-type resins for their proposed reprocessing process, showed that Ru(III) was separated from U(VI) and other elements in a chromatography experiment using 0.5 M HCl [Citation24]. Because the distribution coefficient of Pd(II) in tertiary pyridine-type resin is larger than that of Ru(III) [Citation14], Pd(II) will adsorb along with Ru(III) during chromatography. In the next step, U(VI) and Pu(IV) are separated from the FPs and structural materials except Mo(VI), Fe(III), and Sn(IV). Mo is a nonradioactive FP, while Fe and Sn are structural materials with low radiation levels. Therefore, contamination by Fe and Sn is not considered a problem during the storage of U and Pu. However, for recycling of U and Pu, Fe and Sn, whose volume is higher than that of the FPs, should be separated. A prospective separation method for these species is currently being developed by the present authors.
Figure 6. Proposed process for separation of U and Pu from hardly soluble nuclear materials.
4. Conclusion
The distribution coefficients of the major elements present in 1F debris in HCl were measured using a benzimidazole-type anion exchange resin embedded in porous silica beads, and the distribution coefficient trends were discussed from the viewpoint of complexation. The complexation of Zr(IV) in HCl was also evaluated in conjunction with the UV–vis absorption spectra. Moreover, the selectivity of AR-01 for U(VI) separation was evaluated by chromatographic analysis using a glass column loaded with this resin. Overall, U(VI) was separated from a majority of the FPs and Zr(IV), which is present in fuel debris in appreciable quantities along with U. On the other hand, some nonradioactive materials were not separated from U(VI). Because Pd(II) and Ru(III) form complexes with the resin and it is difficult to elute these species, they should be separated prior to U separation. A U and Pu separation process involving the use of this ion exchange method along with chlorination of hardly soluble nuclear materials by using APNTP was proposed. The principle of this process has been verified. This process may be effective for stabilizing 1F debris and simplifying the material control and accounting process.
Acknowledgments
This work was partially supported by a grant-in-aid for Scientific Research (B) under KAKENHI [grant number 23360423]. The present authors would like to thank Professor emeritus Y. Fujii for his expert advice.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Mid-and-long-term roadmap towards the decommissioning of Fukushima daiichi nuclear power units 1-4 [Internet]. Tokyo: Nuclear Emergency Response Headquarters [cited 2015 Aug 6]. Available from: http://www.meti.go.jp/earthquake/nuclear/decommissioning/committee/fukushimahyougikai/2015/pdf/150615_01g.pdf.
- Akers DW, Carlson ER, Cook BA, et al. TMI-2 core debris grab samples examination and analysis: Part 1. Idaho Falls (ID): EG&G Idaho, Inc.; 1986. (Report GEND-INF-075-Pt.1).
- Akers DW, Carlson ER, Cook BA, et al. TMI-2 core debris grab samples examination and analysis: Part 2. Idaho Falls (ID): EG&G Idaho, Inc.; 1986. (Report GEND-INF-075-Pt.2).
- Ikeuchi H, Kondo Y, Noguchi Y, et al. Suggestion of typical phases of in-vessel fuel-debris by thermodynamic calculation for decommissioning technology of Fukushima-daiichi nuclear power station. Proceedings of the Global 2013 Nuclear Energy at a Crossroad; 2013 Sep 29-Oct 3; Salt Lake City (UT). LaGrand Park (IL): American Nuclear Society; 2013.
- The Evaluation Status of Reactor Core Damage at Fukushima Daiichi Nuclear Power Station Units 1 to 3 [Internet]. Tokyo: Tokyo Electric Power Company [cited 2015 Aug 6]. Available from: http://www.tepco.co.jp/nu/fukushima-np/images/handouts_111130_09-j.pdf.
- Kitagaki T, Suzuki T, Kaneshiki T, et al. Application of atmospheric-pressure non-thermal plasma to chlorination of hardly soluble materials. Prog Nucl Energy. 2014;82:122–125.
- Nogami M, Fujii Y, Sugo T. Radiation resistance type anion exchange resins for spent fuel treatment. J Radioanal Nucl Chem. 1996;203(1):109–117.
- Wei YZ, Kumagai M, Takashima Y, et al. The application of an advanced ion exchange process to reprocessing spent nuclear fuels, (I). J Nucl Sci Technol. 1998; 35(5):357–364.
- Seko M, Takeda K, Onitsuka H, et al. Theoretical consideration and recent progress of chemical enrichment process for uranium enrichment. J Nucl Sci Technol. 1990; 27(11):983–995.
- Kumagai M, Yamaguchi M, Takashima Y, et al. Reprocessing of spent nuclear fuels with a newly developed ion exchange process. Proceedings of the IMechE 1992; 1992 Nov 17–18; London: Mechanical Engineering Publications Limited; 1992.
- Fujii Y, Higuchi N, Haruno Y, et al. Temperature dependence of isotope effects in uranium chemical exchange reactions. J Nucl Sci Technol. 2006; 43(4):400–406.
- Tachibana Y, Yamazaki Y, Nomura M, et al. Molybdenum isotope fractionation in ion exchange reaction by using anion exchange chromatography. J Radioanal Nucl Chem. 2015;303(2):1429–1434.
- Nishihara K, Iwamoto D, Suyama K. Estimation of fuel compositions in Fukushima-Daiichi nuclear power plant (Japan). Ibaraki: Japan Atomic Energy Agency; 2012. (JAEA-Data/Code 2012-018).
- Nogami M, Aida M, Fujii Y, et al. Ion-exchange selectivity of tertiary pyridine-type anion-exchange resin for treatment of spent nuclear fuels. Nucl Technol. 1996;115:293–297.
- Grenthe I, Fuger J, Koning RJM, et al. Chemical thermodynamics of uranium. Issy-les-Moulineaux: OECD NEA Data Bank; 2004.
- Högfeldt E. Stability constants of metal-ion complexes part A: inorganic ligands. Oxford: Pergamon Press; 1982.
- Gamsjäger H, Gajda T, Sangster J, et al. Chemical thermodynamics volume 12: chemical thermodynamics of tin. Issy-les-Moulineaux: OECD Publications; 2012.
- Brown PL, Curti E, Grambow B, et al. Chemical thermodynamics volume 8: chemical thermodynamics of zirconium. Issy-les-Moulineaux: OECD Nuclear Energy Agency, Data Bank; 2005.
- Ogawa N, Sakane H, Miyanaga T, et al. An extended X-ray absorption fine structure analysis of the local structure for zirconium(IV) in acidic aqueous solution [in Japanese]. Bunseki Kagaku. 1986;35:785–788.
- Takasaki F, Ogawa N, Watanabe I, et al. Structure of zirconium(IV) in aqueous zirconium chloride solutions as studied by Zr-K edge extended X-ray absorption fine structure analysis [in Japanese]. Bunseki Kagaku. 2010; 59(6):447–454.
- Arai T, Takeda K, Wei YZ, et al. Ion exchange with chemical reaction: Platinum triad group elements on anion exchange. J Ion Exch. 1995;6(3)90–107.
- Sillén LG, Martell AE, Bjerrum J. Stability constants of metal ion complexes: special publication no.17. London: The Chemical Society, 1964.
- Kofuji H, Watanabe S, Goto I, et al. Basic research programs for the next generation vitrification technology (19) Vitrification properties of MA adsorbents [in Japanese]. Proceedings of the 2015 Fall Meeting of AESJ; 2015 Sep 9–11; Tokyo (Japan): Atomic Energy Society of Japan; 2015.
- Koyama S, Ozawa M, Suzuki T, et al. Development of a multi-functional reprocessing process based on ion-exchange method by using tertiary pyridine-type resin. J Nucl Sci Technol. 2006;43(6):681–689.