
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Cesium was recovered from soil samples obtained in Fukushima prefecture. Isotopic analysis of Cs was performed by γ-spectrometry to determine the activity ratio of 134Cs/137Cs and thermal ionization mass spectrometry was used to determine the isotopic ratios of 133Cs/137Cs and 135Cs/137Cs. The analytical results showed that both the activity ratio of 134Cs/137Cs and the isotopic ratio of 135Cs/137Cs were within the expected values for the Fukushima Daiichi Nuclear Power Plant estimated using the ORIGEN-II code, suggesting that most of the radioactive Cs in the soil sample originated from the Fukushima Daiichi Nuclear Power Plant. The concentration of 137Cs and the contribution of radioactive Cs from global fallout were correlated to the distance from the Fukushima Daiichi Nuclear Power Plant, while the contribution of radioactive Cs from each reactor did not show any similar distance dependence.
1. Introduction
The underexploited applicability of mass spectrometry to analysis of the radioactive Cs released from the accident site of Fukushima Daiichi Nuclear Power Plant (FDNPP) has been emphasized [Citation1–7], although almost all existing studies have utilized γ-spectrometry. The main advantage of mass spectrometry is that 135Cs having a half-life (T1/2) of 2.3 × 106 y is detectable in addition to 134Cs (T1/2 = 2.06 y) and 137Cs (T1/2 = 30.2 y). Mass spectrometry has been used for: (Equation1(1)
(1) ) two-dimensional (2D) analysis of radioactive Cs by monitoring the 134Cs/137Cs and 135Cs/137Cs ratios until the signal of 134Cs became lower than the detection limit [Citation2–4], and (Equation2
(2)
(2) ) source analysis of radioactive Cs from nuclear weapons test [Citation7–9] and from nuclear facilities [Citation10–13].
The inceptive application of mass spectrometry to the analysis of radioactive Cs was performed by Lee and coworkers [Citation8], who detected 135Cs from global fallout (GF) using thermal ionization mass spectrometry (TIMS). Subsequently, mass spectrometric analysis of Cs was performed using inductively coupled plasma mass spectrometry (ICP-MS) [Citation11,Citation12] and resonance ionization mass spectrometry [Citation14,Citation15] for the analysis of radioactive Cs in irradiated nuclear fuel [Citation16,Citation17] and environmental sample obtained in/near nuclear facilities [Citation10–12].
The application of mass spectrometry to the analysis of radioactive Cs released from the accident at FDNPP has been intensively studied by Shibahara et al. [Citation2–4] and others [Citation1,Citation5,Citation6]. They analyzed the isotopic composition of Cs in rainwater, plant samples, and soil samples: rainwater and soil samples were analyzed by ICP-MS [Citation1,Citation5,Citation6], and plant samples were analyzed by TIMS [Citation2–4]. These results showed that the 135Cs/137Cs isotopic ratio in the environmental samples obtained in/around Fukushima prefecture was different from that of GF and the Chernobyl Nuclear Power Plant (CNPP). In addition, Shibahara et al. determined the 134Cs/137Cs activity ratio in plant samples using TIMS, suggesting that 2D analysis of radioactive Cs was useful for source analysis [Citation2–4].
We previously analyzed the isotopic composition of nuclear fuel materials [Citation18,Citation19] and fission products [Citation2–4,Citation20] released in the accident at FDNPP for safety assessment and source analysis of radioactive nuclides. In the previous studies, we analyzed the radioactive Cs in plant samples obtained from the high dose rate area in Fukushima prefecture (>12 μSv/h) [Citation2], and compared the analytical result for Cs with the isotopic composition of Sr [Citation3,Citation4]. However, the high dose rate area is a small portion of the contaminated area in Fukushima prefecture. In this study, we analyze the isotopic composition of radioactive Cs in soil samples obtained from the expanded sampling area (>0.6 μSv/h) along with the high dose rate area in order to clarify the variation of the 135Cs/137Cs isotopic ratio and the 134Cs/137Cs activity ratio for source analysis of radioactive Cs.
2. Experimental
2.1. Sample preparation
The soil samples listed in were obtained from the public area in Fukushima prefecture: the concentration of 137Cs ranged from 3.2 × 10−1 to 1.2 × 104 Bq/g. After drying at 373 K, about 5 g of soil sample screened through a 2 mm mesh screen was immersed in 8 M HNO3 (TAMAPURE-AA 100) and heated at 403 K. The dried samples were again immersed in 8 M HNO3 and were centrifuged to remove the insoluble residues. After centrifugation, H2O2 (TAMAPURE-AA 100) was added to prepare an 8 M HNO3/0.3% H2O2 sample solution for isolation of the transuranic (TRU) elements such as U and Pu via extraction chromatography using TRU-resin™ [Citation18,Citation19].
Table 1. List of soil samples analyzed in this study.
2.2. Recovery of Cs
The sample solution was adjusted to a concentration of 4 M HNO3 to recover Sr using Sr-resin™ [Citation2–4] after isolation of the TRU elements [Citation18,Citation19]. The eluent from the Sr-resin was used for recovery of Cs with ammonium phosphomolybdate (AMP).
A 0.1 g portion of AMP was added to the 4 M HNO3 solution eluted from the Sr-resin containing Cs. After stirring for a few hours, the supernatant was removed by centrifugation. A 20 mL aliquot of 2 M ammonium hydroxide solution was used to dissolve the residue for anion-exchange chromatography. A column (54 mm in length and 6.5 mm in diameter) was filled to 3 mL with anion-exchange resin (DOWEX™ 1 × 8; 100–200 mesh). After the final conditioning, the ammonium hydroxide Cs solution was added to the column. The eluent from the column was collected and heated to dryness. The residue was dissolved in 20 mL of 0.1 M HNO3 for further purification of Cs with AMP. The process of purification of Cs with AMP and anion-exchange chromatography was carried out three times (hereafter termed the ‘three-time method’: first time: 4 M HNO3 system, second and third time: 0.1 M HNO3 system). Based on results presented by Yamagata [Citation23], this method should give a recovery ratio of ca. 86%. After the third purification of Cs with AMP and anion-exchange chromatography, 20 mL of 0.1 M HNO3 Cs solution was obtained for the final purification of Cs by the cation-exchange chromatography.
The cation-exchange resin (DOWEX™ 50WX8; 100–200 mesh) was loaded into a column (54 mm in length and 6.5 mm in diameter) to 3 mL. After the final conditioning with 0.1 M HNO3, the Cs solution was loaded into the column and the resin was rinsed with 0.1 M HNO3. Cs was recovered from the resin by washing with 20 mL of 1.5 M HCl (TAMAPURE-AA 100). The eluent was heated to dryness, and the residue was dissolved in 1 M HNO3 solution for analysis of the isotopic composition of Cs.
2.3. Analysis of isotopic composition of Cs
The radioactive Cs (134Cs and 137Cs) was quantified by γ-spectrometry, and the 135Cs/137Cs and 133Cs/137Cs isotopic ratios were measured by TIMS (TRITON™ T1, Thermo Fisher Scientific) with a single filament using a TaO method; it was confirmed that this method produces a Cs ion beam of high intensity and high stability, and does not generate isobaric interference by species such as Ba etc., as encountered in our previous study [Citation2]. A several μL aliquot of each sample solution was loaded onto a rhenium filament with a TaO activator [Citation2–4,Citation24]; the amount of loaded 137Cs ranged from 1 to 5 Bq. Measurement of the isotopic ratios of Cs was carried out in the same manner used in a previous study [Citation2–4], and the mass spectrum was obtained with a secondary electron multiplier (SEM) detector because of the low total amounts of radioactive Cs loaded on the filament.
The IAEA-156 [Citation21] and IAEA-373 [Citation22] standard materials were used as reference materials. About 2.5 g of the standard materials was employed, and the same method used for the recovery of Cs (described above) was utilized.
3. Results and discussion
3.1. Results of Cs analysis by γ-spectrometry and TIMS
The 134Cs/137Cs activity ratio determined by γ-spectrometry and the 135Cs/137Cs and 133Cs/137Cs isotopic ratios determined by TIMS are summarized in . also summarizes the results of isotopic analysis of the radioactive Cs recovered from plant samples [Citation2–4]. In addition to using γ-spectrometry, the 134Cs/137Cs isotopic ratio of the plant samples was also determined by TIMS in the previous study. This is because the amount of natural Cs (133Cs) in the samples was low enough for detection of the 134Cs signal. In the present study, this was not applicable. Since the soil samples contain a larger amount of natural Cs than radioactive Cs, interference due to tailing of the 133Cs ion beam into the 134Cs ion beam occurs. The average and median 133Cs/137Cs isotopic ratios of the soil samples were 4.1 × 106 and 5.2 × 105, respectively (the corresponding predicted 134Cs/133Cs isotopic ratios were 8.2 × 10−8 and 1.0 × 10−7 since the 134Cs/137Cs isotopic ratio was ca. 0.02 at Nov. 2015). These values are significantly higher than those of the plant samples, as shown in . The 135Cs/137Cs isotopic ratio of IAEA-156 obtained in this study was in agreement with that of IAEA-373 obtained in this study and the reported data for IAEA-156 [Citation25], within error; these values were used as the 135Cs/137Cs isotopic ratio of the nuclear material released in the accident at CNPP.
Table 2. Analytical results of activity ratio and isotopic ratio of soil samples.
The average analytical error was ∼0.1% for the 135Cs/137Cs isotopic ratio determined in a previous analysis of Cs in plant samples [Citation2–4], while the analytical errors for the 135Cs/137Cs ratio in this study was higher than that of the previous study. The errors in the 135Cs/137Cs isotopic ratio analysis of the soil samples were compared with the concentrations of 137Cs in the soil samples as shown in . The errors for the plant samples are also plotted in . The plot shows that the error was correlated with the concentration of 137Cs. In the reports by Snow et al. [Citation25–27], the error in the 135Cs/137Cs isotopic ratio was not influenced by the loading amount in the range of 1–5 Bq. The error obtained in this study might not be strongly influenced by the Cs loading based on this result ( shows the dependence of the error on the concentration of 137Cs); the results presented by Snow et al. also showed a good correlation with this dependency.
Figure 1. Error in 135Cs/137Cs ratio measurement as a function of concentration of 137Cs in soil. Closed circles are results of analysis of soil sample in this study. Closed diamonds are that of plant standard material of IAEA-156 and IAEA-373 in this study. Closed squares are that of plant sample in previous study [Citation2–4]. Open circles and open squares are the soil and plant samples of standard material reported by Snow [Citation25,Citation27]: averaged values of errors were used for this plot.
![Figure 1. Error in 135Cs/137Cs ratio measurement as a function of concentration of 137Cs in soil. Closed circles are results of analysis of soil sample in this study. Closed diamonds are that of plant standard material of IAEA-156 and IAEA-373 in this study. Closed squares are that of plant sample in previous study [Citation2–4]. Open circles and open squares are the soil and plant samples of standard material reported by Snow [Citation25,Citation27]: averaged values of errors were used for this plot.](/cms/asset/3790d4c9-e373-4696-879a-1653c894ec8c/tnst_a_1223560_f0001_b.gif)
The Cs recovery scheme used in the previous study consisted of the adsorption of Cs by AMP, anion-exchange chromatography, and cation-exchange chromatography (hereafter, ‘one-time method’) [Citation2–4]. It was confirmed that the three-time method was required for the recovery of Cs from the soil samples in this study (except for samples OK01 and OK03) and the IAEA-156 and IAEA-373 standard reference materials. In the case of the one-time method, the observed mass spectra were influenced by impurities, as also observed by Snow and coworkers [Citation25]. However, in the previous study [Citation2–4], the analytical error for the plant sample data showed a weaker dependence on the concentration of 137Cs; the magnitude of the error was also lower than that of the soil sample. This suggests that the error was influenced by the type of sample. Such variations may be caused by the matrix effect of the sample, suggesting there could be a better method for the recovery of Cs.
3.2. Comparison of analytical results for radioactive Cs based on sampling location
The sampling locations, the 134Cs/137Cs activity ratio, and the 135Cs/137Cs and 133Cs/137Cs isotopic ratios are shown in . The OK01, OK02, OK03, OK04, FT01, FT02, NM02, and IT01 (group-1) samples, which were obtained from the high dose rate area (> 12 μSv/h), had identical isotopic 135Cs/137Cs ratios within error; the average 135Cs/137Cs isotopic ratio was 0.364. Although the 134Cs/137Cs activity ratio of these samples was ca. 1, some variation in the 134Cs/137Cs activity ratio was also found among the samples; the minimum value was 0.977, the maximum value was 1.104, and the average value was 1.015. These samples have a high 137Cs concentration and low 133Cs/137Cs isotopic ratio on the whole; the 137Cs concentration ranged from 3.9 Bq/g to 1.2 × 104 Bq/g, and the 133Cs/137Cs isotopic ratio ranged from 7.0 × 102 to 5.7 × 105. The KM01 sample (the concentration of 137Cs was 5.2 Bq/g, and the 133Cs/137Cs isotopic ratio was 5.2×105) may also be included in this group since the 135Cs/137Cs isotopic ratio and the 134Cs/137Cs activity ratio had similar values. The main source of radioactive Cs in these samples would be FDNPP (it was confirmed that the OK01, OK04, FT01, and FT02 samples were also contaminated with Pu from FDNPP [Citation19]). The other samples were classified into two groups: (Equation1(1)
(1) ) only the isotopic ratio 135Cs/137Cs was higher than the average value for the high dose rate area (samples NH01 and KU01 obtained from the south side of the high dose rate area and samples MS02 and IT02 obtained from the north side of the high dose rate area; group-2-a and group-2-b, respectively), and (Equation2
(2)
(2) ) the 134Cs/137Cs activity ratio was lower than the average value for the high dose rate area and the 135Cs/137Cs isotopic ratio was higher than the average value for the high dose rate area (samples NM01 and MS01 obtained from the north side of FDNPP; group-3). These two sample groups had a relatively low concentration of 137Cs (ranging from 3.2×10−1 Bq/g to 9.6 Bq/g) and a relatively high 133Cs/137Cs isotopic ratio (ranging from 5.3×106 to 2.7×107).
Figure 2. Sampling areas and results of measurement of isotopic ratio and activity ratio. These areas are same as them of previous study reporting isotopic ratios of Pu and U: area showing boxed characters means that contamination of Pu from FDNPP was observed in previous study [Citation19]. Contour of radiation dose rate were made using published data from MEXT [Citation28].
![Figure 2. Sampling areas and results of measurement of isotopic ratio and activity ratio. These areas are same as them of previous study reporting isotopic ratios of Pu and U: area showing boxed characters means that contamination of Pu from FDNPP was observed in previous study [Citation19]. Contour of radiation dose rate were made using published data from MEXT [Citation28].](/cms/asset/8e91194e-dfd5-4afb-839a-cb35efd67bc6/tnst_a_1223560_f0002_b.gif)
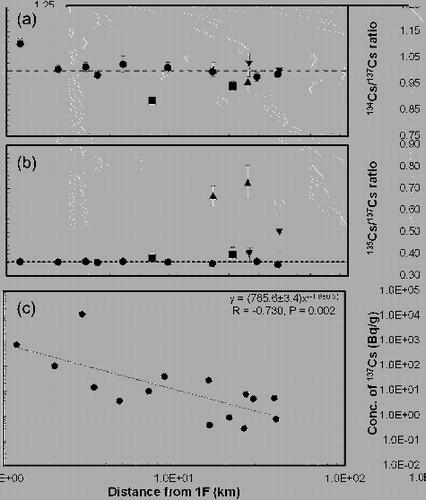
The relationships between the distance from FDNPP to the sampling area and the 134Cs/137Cs activity ratio, the 135Cs/137Cs isotopic ratio, and the concentration of 137Cs are presented in . The variation of the 134Cs/137Cs activity ratio was significant at shorter distances and this variation was also observed for all groups as above mentioned (especially, group-1 and group-3), although the 134Cs/137Cs activity ratios were around unity. The 135Cs/137Cs isotopic ratio for the samples in group-1 were in agreement with each other in moving from the shorter distance to the longer distance, while that of group-2 showed the characteristic variation from the middle distance (group-2-a) to the longer distance (group-2-b). The concentration of 137Cs was negatively correlated with the distance between the sampling area and FDNPP on the whole, and the concentration of 137Cs did not show the characteristic profile depending on the sample group as mentioned above.
Figure 3. Activity ratio of 134Cs/137Cs (a), isotopic ratio of 135Cs/137Cs (b) and concentration of 137Cs (c) as a function of distance among FDNPP and sampling area. Closed circle, closed triangle, closed inverted triangle and closed square are classified into group-1, group-2-a, group-2-b and group-3, respectively.
3.3. Evaluation of contribution of radioactive Cs-1: FDNPP vs. GF
For detailed evaluation of radioactive Cs in the soil sample, the 134Cs/137Cs activity ratio was compared with the 135Cs/137Cs isotopic ratio, as shown in . Both the 134Cs/137Cs activity ratio and the 135Cs/137Cs isotopic ratio of radioactive Cs from each reactor presented in were calculated using the ORIGEN-II code [Citation29]; the reported 134Cs/137Cs activity ratio of polluted water [Citation30] is also plotted in this figure. Both the 134Cs/137Cs activity ratio and the 135Cs/137Cs isotopic ratio observed in this study are close to the values for FDNPP calculated using the ORIGEN-II code, meaning that the radioactive Cs in the soil samples mainly originated by from the accident at FDNPP ((a)). The samples in group-1 obtained from the high dose rate area showed a distribution centered in the middle point between reactors 1 and 2, sample K01 being an exception ((b)). This suggests that the contribution from reactor 2 would be the major source of radioactive Cs in sample of OK01, and the radioactive Cs in the other samples from group-1 would be evenly derived from reactors 1 and 2 (and also reactor 3). The samples from group-2, having a higher 135Cs/137Cs isotopic ratio than that of group-1, were located on the line between FDNPP and GF. This would be caused by mixing of the radioactive Cs form GF and FDNPP. Comparison between group-2-a and group-2-b shows that group-2-a was located on the line between reactor 2 and GF, and group-2-b was located on the line between the averaged values estimated from the ORIGEN-II code and GF calculation. Group-3, which was obtained from the middle dose rate area, was located around reactor 1, suggesting correlation between this group and the radioactive Cs from reactor 1 of FDNPP.
Figure 4. 135Cs/137Cs (atomic ratio) vs. 134Cs/137Cs (activity ratio) observed in this study. Closed circle, closed triangle, closed inverted triangle and closed square are classified in group-1, group-2-a, group-2-b and group-3, respectively. Open circle of FDNPP represents calculation results from estimation of radioactive nuclides with ORIGEN-II code [Citation29]. Open square is estimation results of GF [Citation7–9]. Closed diamond means CNPP observed in this study. Colored bar represents values reported for 134Cs/137Cs activity ratio in polluted water [Citation30].
![Figure 4. 135Cs/137Cs (atomic ratio) vs. 134Cs/137Cs (activity ratio) observed in this study. Closed circle, closed triangle, closed inverted triangle and closed square are classified in group-1, group-2-a, group-2-b and group-3, respectively. Open circle of FDNPP represents calculation results from estimation of radioactive nuclides with ORIGEN-II code [Citation29]. Open square is estimation results of GF [Citation7–9]. Closed diamond means CNPP observed in this study. Colored bar represents values reported for 134Cs/137Cs activity ratio in polluted water [Citation30].](/cms/asset/9da7f9b1-c60c-44d5-8c5a-f283a6a97b52/tnst_a_1223560_f0004_b.gif)
Similar to the previous analysis of Pu [Citation19], the contribution of the radioactive Cs from GF was evaluated by regression analysis using the following mixing equations;
(1)
(1)
(2)
(2) where x, f, and g indicate the fraction of radioactive Cs from GF in the sample, the 134Cs/137Cs activity ratio of the sample, and the 135Cs/137Cs isotopic ratio of the sample, respectively. The suffixes GF and FDNPP indicate the radioactive Cs from GF and FDNPP. By using EquationEquations (1)
(1)
(1) and (Equation2
(2)
(2) ), regression analysis of the values for FDNPP and GF was performed for rough estimation of the contribution of the radioactive Cs from GF to the samples (). For the values of [134Cs/137Cs]FDNPP and [135Cs/137Cs]FDNPP, the average value for the high dose area were used, and 0 and 3.0 were used for the values of [134Cs/137Cs]GF and [135Cs/137Cs]GF. Eight samples were found to be influenced by the radioactive Cs from GF based on the regression analysis. The samples of group-2 and group-3 showed the effects of radioactive Cs from GF. The contribution of the radioactive Cs from GF ranged from 1.0% to 12.8%. The samples from group-1 showed no significant effect from the radioactive Cs from GF, except for samples FT01 and IT01.
Table 3. Result of evaluation of contribution of Cs from GF and reactor 1 by using EquationEquations (1)(1)
(1) –(Equation2
(2)
(2) ) and EquationEquations (5)
(5)
(5) –(Equation6
(6)
(6) ).
Comparison of the concentration of 137Cs in the soil and the contribution of radioactive Cs from GF showed an inverse dependence, as shown in (a), and this dependence is described by the following correlation equation:
(3)
(3) where h indicates the contribution of radioactive Cs from GF. This equation suggests that the contribution of radioactive Cs from GF would be 100% for 137Cs concentrations in the soil of 6.1 mBq/g to 48.1 mBq/g (the medium value was estimated as 25.6 mBq/g). This range shows agreement with the general concentration of 137Cs in soil in Japan before the accident at FDNPP. The reported range was from 0.5 mBq/g to 34.9 mBq/g (the average presented in Ref. [Citation31] was 14.1 mBq/g, and the range presented in Ref. [Citation32] was 0.7 mBq/g to 42.7 mBq/g).
Figure 5. Contribution of 137Cs of GF (a), isotopic ratio of 133Cs/137Cs (b) and that of reactor 1 (c) as a function of concentration of 137Cs. Closed circle, closed triangle, closed inverted triangle and closed square are classified in group-1, group-2-a, group-2-b and group-3, respectively. Open circle in (a) was evaluated that the contribution of radioactive Cs of GF in these samples were negligible small (less than 0.1%).
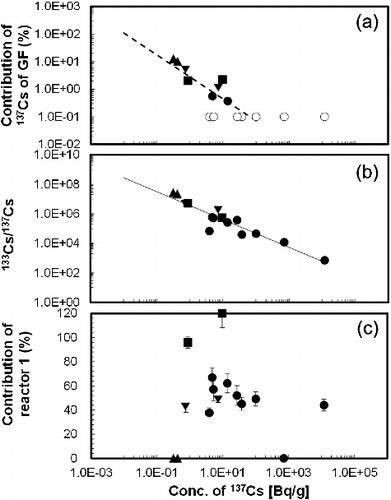
The 133Cs/137Cs isotopic ratios presented in were also inversely correlated with the concentration of 137Cs ( (b)), and this correlation gave rise to the following correlation equation:
(4)
(4) where i indicates the 133Cs/137Cs isotopic ratio in the soil sample. According to this correlation, the concentration of 133Cs could be estimated as ranging from 0.5 ppm to 1.6 ppm (the median value was estimated as 0.9 ppm). This range also shows agreement with the general concentration of 133Cs in soil, i.e. 1ppm to 4 ppm in the crust [Citation33], and 0.1 ppm to several tens of ppm (the mean was around 5 ppm) in Japanese soil [Citation34]. The dissolution of radioactive Cs from the soil sample would follow the same trend as that of 133Cs according to EquationEquations (3)
(3)
(3) and (Equation4
(4)
(4) ), provided that the soil sample was not in a disturbed location such as the JSAC soil sample [Citation27], and the radioactive Cs in the soil sample would exist in essentially the same form as natural Cs (namely 133Cs). Since the concentration of 137Cs was also correlated with the distance between the sampling area and FDNPP, both the contribution of the radioactive Cs of from GF and the 133Cs/137Cs isotopic ratio would show a relationship with the distance between the sampling area and FDNPP.
3.4. Evaluation of contribution of radioactive Cs-2: Source term evaluation
Similar to the previous study discussing the contribution of radioactive Cs from each reactor to the contaminated plant samples [Citation2–4] and evaluation of the contribution of the radioactive Cs from GF mentioned above, the contribution of the radioactive Cs from each reactor is discussed herein. It is recognized that there were several events of radioactive Cs release in the accident at FDNPP, and there was some extent of bias in the contribution of radioactive Cs from each reactor in these events [Citation35–37]. Nishihara et al. reported that the radioactive Cs in each reactor would be reflected in the variation of the 134Cs/137Cs activity ratio and the 135Cs/137Cs isotopic ratio [Citation29], suggesting that the source of the radioactive Cs in the environmental samples could possibly be evaluated from the isotopic data such as the isotopic ratio and the activity ratio of radioactive Cs.
Evaluation of the contribution of radioactive Cs from each reactor in FDNPP to environmental samples has been attempted in our previous study [Citation2–4] and in studies by other groups [Citation5,Citation6,Citation27]; Zheng et al. concluded that the largest contributor to radioactive Cs in environmental samples originated from reactor 2, and Snow et al. showed that the contribution of radioactive Cs from each reactor to the environmental samples varied with the sample and the sampling location. Snow et al. also discussed the release timing of radioactive Cs (the radioactive Cs was released before/after significant core damage [Citation27] because of the variation in the isotopic ratio of 135Cs/137Cs as a function of the fuel rod height).
Based on the report by Snow et al., the radioactive Cs in the plant samples assayed in our previous study would originate from: (Equation1(1)
(1) ) combined contribution from reactors 1, 2, and 3 after major core damage to each reactor, or (Equation2
(2)
(2) ) only melting/cladding failure of the tops of the fuel rods in reactor 2 prior to March 15 (the expected timing of the primary emissions of radioactive Cs from reactor 2) [Citation27]. In this study, the combined contribution from reactors 1, 2, and 3 was focused on because of (Equation1
(1)
(1) ) the results of evaluation of the core using MAAP and MELCOR [Citation38], (Equation2
(2)
(2) ) the results of monitoring the radiation dose near FDNPP [Citation37], and (Equation3
(3)
(3) ) mixing of the radioactive Cs in the fuel rod (the integrated isotopic ratio for the top of the fuel rod and at a height of 100 cm from the top should agree with the averaged isotopic ratio, within error, according to the data reported by Snow and coworkers [Citation27]).
For evaluation of the radioactive Cs from each reactor, three mixing equations (such as EquationEquations (1)(1)
(1) and (Equation2
(2)
(2) )) are necessary, since there are three reactors in FDNPP. However, we could obtain only two mixing equations for the 134Cs/137Cs activity ratio and the 135Cs/137Cs isotopic ratio. From the calculation data reported by Nishihara and coworkers [Citation29], the 134Cs/137Cs and 135Cs/137Cs activity ratio would follow the order: [134Cs/137Cs]reactor 1 < [134Cs/137Cs]reactor 3 < [134Cs/137Cs]reactor 2, and [135Cs/137Cs]reactor 1 > [135Cs/137Cs]reactor 3 > [135Cs/137Cs]reactor 2. Both the 134Cs/137Cs activity ratio and the 135Cs/137Cs isotopic ratio for most of the group-1 samples (obtained in the high dose area) were in agreement with those of the plant samples, suggesting that the contamination in these samples originated from a mixture of the radioactive Cs from reactors 1, 2, and 3, in accordance with the report by Snow [Citation27]. Thus, the degree of bias for the activity ratio and the isotopic ratio of reactor 1/reactor 2 from the combined contributions of reactors 1, 2, and 3 were evaluated.
Similar to the evaluation of the contribution of the radioactive Cs from GF, the contribution of the radioactive Cs from reactor 1 versus reactor 2 was evaluated by using the following equations;
(5)
(5)
(6)
(6) where y, j, and k indicate the fraction of radioactive Cs from reactor 1 in the sample, the 134Cs/137Cs activity ratio of the sample and the 135Cs/137Cs isotopic ratio of the sample, respectively. By using EquationEquations (5)
(5)
(5) and (Equation6
(6)
(6) ), the 134Cs/137Cs activity ratios from reactors 1 and 2 calculated using ORIGEN-II code [Citation29] and the 135Cs/137C isotopic ratios were analyzed using regression analysis for estimation of the contribution of radioactive Cs from reactor 1 to the samples (). In this regression analysis, the error in the results due to ORIGEN-II code was not discussed since the error was not reported [Citation29]. Same as the variation in both the 134Cs/137Cs activity ratio and the 135Cs/137Cs isotopic ratio as a function of the fuel rod height as mentioned above, these values will also show the variation depending on the radial position in the reactor core since the reactor core consists of several batch of fuel assembly (the results due to ORIGEN-II code [Citation29] was discussed about the effect of batch and was summarized as the averaged values). The bias of the isotopic data due to the batch of fuel assembly was not discussed in this study, since (Equation1
(1)
(1) ) the variation of the isotopic data due to the batch was not reported [Citation29], and (Equation2
(2)
(2) ) the combined contribution from each reactor after major core damage as mentioned above was focused.
The estimated data for the group-1 samples (except for OK01) ranged from 37.1% to 66.9% (av. 51.7±9.8%). These values are in agreement with the results for group-2-b, suggesting that the samples from group-2-b were also contaminated with a mixture of radioactive Cs from reactors 1, 2, and 3. The results for the other samples showed a significant bias for reactor 1/reactor 2. The group-2-a samples and the OK01 sample obtained from the south side of the high dose rate area showed an influence from reactor 2, while the group-3 samples showed an influence from reactor 1.
In contrast with the contribution of radioactive Cs from GF and the 133Cs/137Cs isotopic ratio ( (a) and (b)), the contribution of radioactive Cs from reactor 1 showed no clear dependence on the concentration of 137Cs ( (c)). The contribution of radioactive Cs from each reactor would not show any dependence on the concentration of 137Cs (or the distance between the sampling area and FDNPP as mentioned above), but would show direction dependence. This means that the isotopic analysis of Cs by TIMS (as well as ICP-MS) may indicate the distribution of radioactive Cs from each reactor in more detail and provide a wider analytical range.
In contrast with the contribution of radioactive Cs from GF and the 133Cs/137Cs isotopic ratio ( (a) and (b)), the contribution of radioactive Cs from reactor 1 shows no clear dependence on the concentration of 137Cs ( (c)). The contribution of radioactive Cs from each reactor would not show any dependence on the concentration of 137Cs (or distance between the sampling area and FDNPP as mentioned above), but would show the direction dependence. This means that the isotopic analysis of Cs by TIMS (as well as ICP-MS) may indicate the distribution of radioactive Cs from each reactor in more detail and provide a wider analytical range.
4. Conclusion
Cesium was recovered from soil samples obtained from Fukushima prefecture. The isotopic analysis of Cs was performed by γ-spectrometry to determine the 134Cs/137Cs activity ratio, and TIMS was applied for analyzing the 133Cs/137Cs and 135Cs/137Cs isotopic ratios. The results of the analysis showed that both the 134Cs/137Cs activity ratio and the 135Cs/137Cs isotopic ratio of the soil samples were within the expected values for FDNPP estimated using ORIGEN-II code, suggesting that most of the radioactive Cs in the soil originated from FDNPP. Regression analysis was used to evaluate the contribution of radioactive Cs from GF and that from reactor 1 of the FDNPP. The contribution of radioactive Cs from GF was correlated with the distance between the sampling area and FDNPP, while that of reactor 1 showed no correlation with the concentration of 137Cs in the soil, but was correlated with the direction from FDNPP. This data should be useful for the analysis of the distribution of the radioactive Cs from each reactor and for the source analysis of radioactive Cs.
Acknowledgments
This work was supported by the KUR Research Program for Scientific Basis of Nuclear Safety.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Ohno T, Muramatsu Y. Determination of radioactive cesium isotope ratios by triple quadrupole ICP-MS and its application to rainwater following the Fukushima Daiichi Nuclear Power Plant Accident. J Anal At Spectrom. 2014;29:347–351.
- Shibahara Y, Kubota T, Fujii T, et al. Analysis of cesium isotope compositions in environmental samples by thermal ionization mass spectrometry -1. A preliminary study for source analysis of radioactive contamination in Fukushima prefecture. J Nucl Sci Technol. 2014;51:575–579.
- Shibahara Y, Kubota T, Fukutani S, et al. Radiological issues for Fukushima's revitalized future. Tokyo (Japan): Springer; 2016. Chapter 4, Application of mass spectrometry for analysis of cesium and strontium in environmental samples obtained in Fukushima prefecture; p. 33–46.
- Shibahara Y, Kubota T, Fukutani S, et al. Application of mass spectrometry for analysis of cesium and strontium in environmental samples obtained in Fukushima prefecture. Fukushima: International Symposium on Radiological Issues for Fukushima's Revitalized Future; 2015.
- Zheng J, Tagami K, Bu W, et al. 135Cs/137Cs isotopic ratio as a new tracer of radiocesium released from the Fukushima Nuclear Accident. Environ Sci Technol. 2014;48:5433–5438.
- Zheng J, Bu W, Tagami K, et al. Determination of 135Cs and 135Cs/137Cs atomic ratio in environmental sample by combining ammonium molybdophosphate (AMP)-selective Cs adsorption and ion-exchange chromatographic separation to triple-quadrupole inductively coupled plasma-mass spectrometry. Anal Chem. 2014;86:7103–7110.
- Russell BC, Croudace IW, Warwick PE. Determination of 135Cs and 137Cs in environmental samples: a review. Anal Chim Acta. 2015;890:7–20.
- Lee T, Ku TL, Lu HL, et al. First detection of fallout Cs-135 and potential applications of 137Cs/135Cs ratios. Geochim Cosmochim Acta. 1993 Jul;57:3493–3493.
- Snyder DC, Delmore JE, Tranter T, et al. Radioactive cesium isotope ratios as a tool for determining dispersal and re-dispersal mechanisms downwind from the Nevada nuclear security site. J Environ Radact. 2012;110:46–52.
- Delmore JE, Snyder DC, Tranter T, et al. Cesium isotope ratios as indicators of nuclear power plant operations. J Environ Radact. 2011;102:1008–1011.
- Russell BC, Warwick PE, Croudace WC. Calixarene-based extraction chromatographic separation of 135Cs and 137Cs environmental and waste samples prior to sector field ICP-MS analysis. Anal Chem. 2014;86:11890–11896.
- Taylor VF, Evans RD, Cornett RJ. Preliminary evaluation of 135Cs/137Cs as a forensic tool for identifying source of radioactive contamination. J Environ Radact. 2008;99:109–118.
- Snow MS, Snyder DC, Clark SB, et al. 137Cs activities and 135Cs/137Cs isotopic ratios from soils at Idaho national laboratory: a case study for contaminant source attribution in the vicinity of nuclear facilities. Environ Sci Technol. 2015;49:2741–2748.
- Karam LR, Pibida L, McMahon CA. Use of resonance ionization mass spectrometry for determination of Cs ratios in solid samples. Appl Rad Isot. 2002;56:369–374.
- Pibida L, MacMahon CA, Busharw BA. Laser resonance ionization mass spectrometry measurements of cesium in nuclear burn-up and sediment samples. Appl Rad Isot. 2004;60:567–570.
- Moreno JMB, Betti M, Nicolaou G. Determination of caesium and its isotopic composition in nuclear samples using isotope dilution-ion chromatography-inductively coupled plasma mass spectrometry. J Anal At Spectrom. 1999;14:875–879.
- Granet M, Nonell A, Favre G, et al. Cs-Ba separation using N2O as a reactant gas in a multiple collector-inductively coupled plasma mass spectrometer collision-reaction cell: application to the measurements of Cs isotopes in spent nuclear fuel samples. Spectro Chim Acta Part B. 2008;63:1309–1314.
- Shibahara Y, Kubota T, Fujii T, et al. 235U/238U isotopic ratio in plant samples from Fukushima Prefecture. J Radioanal Nucl Chem. 2014;303:1421–1424.
- Shibahara Y, Kubota T, Fujii T, et al. Determination of isotopic ratios of plutonium and uranium in soil samples by thermal ionization mass spectrometry. J Radioanal Nucl Chem. 2015;307:2281–2287.
- Kubota T, Shibahara Y, Fukutani S, et al. Cherenkov counting of 90Sr and 90Y in bark and leaf samples collected around Fukushima Daiichi Nuclear Power Plant. J Radioanal Nucl Chem. 2015;303:39–46.
- International Atomic Energy Agency. Reference sheet of IAEA-156. Vienna: IAEA; 1990.
- Strachnov V, Larosa J, Dekner R, et al. Report on the intercomparison run IAEA-373: determination of radionuclides in grass sample IAEA-373. Vienna: IAEA; 1996.
- Yamagata N, Iwashima K, Tajima E. Determination of rubidium and cesium by neutron activation followed by gamma spectrometry. J Atomic Energy Soc Jpn. 1962 Aug;4:534–539.
- Birck JL. Precision K-Rb-Sr isotopic analysis: application to Rb-Sr chronology. Chem Geol. 1986;56:73–83.
- Snow MS, Snyder DC. 135Cs/137Cs isotopic composition of environmental samples across Europe: environmental transport and source term emission applications. J Environ Radact. 2016;151:258–263.
- Snow MS, Snyder DC, Mann NR, et al. Method for ultra-trace cesium isotope ratio measurements from environmental samples using thermal ionization mass spectrometry. Int J Mass Spescrom. 2015;381–382:17–24.
- Snow MS, Snyder DC, Delmore JE. Fukushima daiichi reactor source term attbitution using cesium isotope ratios from contaminated environmental samples. Rapid Commun Mass Spectrom. 2016;30 523–532.
- Ministry of Education, Culture, Sports, Science and Technology. Analytical results of radionuclide in soil. Fukushima: MEXT; 2011. Japanese.
- Nishihara K, Iwamoto H, Suyama K. [Estimation of fuel compositions in Fukushima-Daiichi Nuclear Power Plant]. Ibaraki: Japan Atomic Energy Agency; 2012, JAEA-Data/Code 2012-018. Japanese.
- Komori M, Shozugawa K, Nogawa N, et al. [Evaluation of radioactive contamination caused by each plant of Fukushima Daiichi Nuclear Power Plant station using 134Cs/137Cs activity ratio as an index]. Bunseki Kagaku. 2013;62:475–483. Japanese.
- Fukuyama T, Onda Y, Gomi T, et al. Quantifying the impact of forest management practice on the runoff of the surface-derived suspended sediment using fallout radionuclides. Hydrol Process. 2010;24:596–607.
- Ministry of Education, Culture, Sports, Science and Technology. Analytical method of radioactive Cs. Fukushima: MEXT; 1976. Japanese. Available from: http://www.kankyo-hoshano.go.jp/series/lib/No3.pdf (accessed on 15 Nov. 2015).
- Burt RO. Kirk-Othmer encyclopedia of chemical technology (4th ed.). New York (NY): John Wiley & Sons; 1993. Cesium and cesium compounds: 749–764.
- Yamasaki S, Takeda A, Nanzyo M, et al. Background levels of trace and ultra-trace elements in soils of Japan. Soil Sci Plant Nutr. 2001;47:755–765.
- Tsuruta H, Oura Y, Ebihara M, et al. First retrieval of hourly atmospheric radionuclides just after the Fukushima accident by analyzing filter-tapes of operational air pollution monitoring stations. Sci Rep. 2014;4:6717.
- Nagai H, Katata G, Terada H, et al. Radiation monitoring and dose estimation of the Fukushima Nuclear Accident. Tokyo (Japan): Springer; 2015. Chapter 15, Source term estimation of 131I and 137Cs discharged from the Fukushima Daiichi Nuclear Power Plant into the atmosphere; p. 155–173.
- Tsuruta H, Nakajima T. [Radioactive materials in the atmosphere released by the accident of the Fukushima Daiichi Nuclear Power Plant]. Chikyukagaku. 2012;46:99–111. Japanese.
- Tanaka S, Kado S. Reflections on the Fukushima Daiichi Nuclear Accident. New York (NY): Springer; 2015. Chapter 3, Analysis of radioactive release from the Fukushima Daiichi Nuclear Power Station; p. 51–83.