
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
We perform first-principles total energy and vibration spectrum calculations to study the effect of Cr/V on the H formation and diffusion at temperature range 300–2100 K in dilute W–Cr/W–V alloy. Temperature and H chemical potential are two important factors to affect the H formation energy and migrating energy. The H formation energy referring to the static/temperature-dependent H chemical potential decreases/increases with the temperature. At each given temperature, the presence of Cr in W reduces the H formation energy, while the existence of V in W has little effect on the H formation energy. The diffusion energy barrier and pre-exponential factor strongly depend on the temperature and increase with the temperature from 300 to 2100 K. Both Cr and V additions in W has a large consequences on H migrating energy. The energy barriers at any given temperature can be reduced by ∼0.05 and 0.10 eV in W–Cr and W–V alloys, respectively. The current study reveals that vibration free energy plays a decisive role in the formation energy and migrating energy of H with the temperature, while the thermal expansion energy has little influence on the H formation energy and migrating energy with the temperature.
1. Introduction
Tungsten (W) is the potential candidates for the first-wall in fusion reactor because W exhibits the low hydrogen (H) sputtering erosion, the high thermal conductivity and high melting temperature [Citation1–Citation4]. Many efforts have been performed to study the behaviors of H isotopes in W [Citation5–Citation22]. In the role of the first-wall, W has to be subjected to high flux H isotopes, which can adsorb on the W surface and further permeate into the bulk and migrating in the bulk. The retention of H isotopes may result in the modification of physical and mechanical behaviors of W. For example, H in W can cause the embrittlement, decrease of mechanical strength and bubble formations.
Adding alloying elements (AEs) has been considered as an important method to improve the basic physical and mechanical properties of W. However, the AEs will inevitably interact with the impurity H from edge plasma so as to possibly result in the change of microstructure and mechanical properties of W material [Citation23]. Correspondingly, the understanding the interaction between impurity H and AE in W is an extreme important work in the current fusion study.
Up to now, several studies including experiments and computational simulations have addressed some of the relevant issues concerning the interactions of H with AEs (solutes) in W. Experimentally, a few works have been carried out to study the effect of the AEs on the H retention in W, which have demonstrated that the AEs can alter obviously the H retention [Citation24–Citation30]. This suggests that the investigation of interaction between H and AE can play a decisive role in the reliable predication of H recycling and retention in W. Theoretically, Kong et al. recently gave the relevant studies of this subject. They have carried out a series of first-principles simulations to study the interaction of H with interstitial AEs in W [Citation31,Citation32]. Later, they extend their study to the investigation of the interaction between H and substitution AEs in W [Citation33]. Among them, the structures and thermodynamics between AEs and H at the interstitial sites and vacancy are discussed in detail using first-principles static calculations. However, there are still many open questions, particularly those which concern the effect of temperature on the interaction between AE and H in the interstitial lattice sites in W. In other words, the temperature effect has been missed on H thermodynamic properties in these theoretical studies. For instance, how is the H formation energy affected by the temperature, i.e. how does the H formation energy changes with the changing temperature. Furthermore, how are the H diffusion energy barrier and pre-exponential factor influenced by the temperature? In previous most calculations all atomic nuclei are treated as point-like classical particles which can lead to inaccuracies in the description of H diffusion including migrating energy barrier and pre-exponential factor where the temperature effect has been negligible, similar to H thermodynamics in W as well as W-based alloys. Here, we will touch these topics from temperature angle, paying close attention to the influence of temperature on H formation and diffusion in W-based alloys.
In this study, we systematically carry out first-principles total energy and vibration spectrum calculations to investigate the influence of temperature on the formation and diffusion behaviors of H in the interstitial positions in W-based alloys. For W-based alloys, we choose vanadium (V) and chromium (Cr) as the model AEs to study the interaction between AE and H in the interstitial lattice by taking into account the temperature effect. Our choice of choosing V and Cr as the AEs is partly because V has been proposed as a possible AE in W with special research emphasis for the applications in a fusion reactor [Citation34], while Cr is generally regarded as the main alloying component in most transition metals. Our findings demonstrate the significant influences of temperature and alloying addition on the formation and diffusion behaviors of H in W, and provide a reasonable interpretation for the larger scatter of the above-predicted results on H formation and diffusion in W-based alloys.
2. Methodology
2.1. Total energy computations of H–W (W-alloy) supercell
For all the energy calculations, we carried out first-principles simulations using the Vienna Ab-initio Simulation Package (VASP) code [Citation35,Citation36] based on density functional theory (DFT). The electron exchange and correlation were treated within the generalized gradient approximation (GGA) using the Pardew–Burke–Ernzerh form [Citation37–Citation39]. The ionic cores were represented by the projector augmented wave potentials [Citation40,Citation41]. During the geometry optimization, we used the 128-atom supercell containing (4 × 4 × 4) unit cells with the lengths of 12.68 Å in the [100], [010], and [001] directions, respectively. The plane wave energy cutoff and k-point density are both tested for each system to reach an energy convergence of 10−3 eV per atom for the electronic minimization. According to a series of convergence test calculations, the energy cutoff of plane wave for W is chosen as 500 eV, while the Brillouin zone of supercell was sampled with the Monkhorst–Pack scheme [Citation42] using a (3 × 3 × 3) k-points mesh. The calculated equilibrium lattice parameter (a) of W lattice is 3.17 Å, which is in good agreement with the corresponding experimental value of 3.16 Å [Citation43]. The Methfessel–Paxton [Citation44] smearing method was used to integrate the Brillouin zone and account for partial occupancies of the metals near the Fermi level with a smearing width of 0.1 eV. During the calculations, the supercell size and atomic positions are relaxed to equilibrium, and energy minimization is converged until the forces on all the atoms are less than 10−3 eVÅ−1.
2.2. Computations of formation energy and concentration of H in W and W-alloy: thermodynamic and statistical model
In W or W-based alloys, the formation energy of H in an interstitial position at the finite-temperature can be attributed to the variation of the Gibbs free energy at the temperature T and pressure P [Citation45],
(1)
(1)
where GHTotal(T, P) and GbulkTotal(T, P) are, respectively, the total Gibbs free energies of the supercell system (W or W-based alloy) with an interstitial H atom and perfect supercell system (W or W-based alloy) at the temperature T and pressure P, and they can be given as
(2)
(2)
where
is the DFT static energy, which is obtained from the current first-principles calculation as the ground-state energy at a given lattice parameter aT. As to the lattice parameter aT, it changes with the increase of temperature and will be clearly explained in Section 3. The term PV can be neglected for solid state systems [Citation45],
is the vibration Helmholtz free energy [Citation45] and in the quasi-harmonic approximation it is written as
(3)
(3)
where ℏωi is the DFT-obtained vibration energy of normal modes, ℏ is Planck's constant divided by 2π, k is the Boltzmann's constant, and 3 N is the total number of vibration modes.
In EquationEquation (1)(1)
(1) , the μH(T, P) is the chemical potential of H, which is related to temperature T and pressure P. The H chemical potential μH(T, P) is generally written as [Citation46–Citation48]
(4)
(4)
where μH(T = 0 K) is half of the energy of an H2 molecule at T = 0 K. The total energy of an H2 molecule is calculated by putting an H2 molecule in a cubic vacuum box with 12.68 Å sides and simultaneously carrying out a zero-point energy (ZPE) calculation at T = 0 K. According to the current calculation, the energies of an H2 molecule without and with ZPE-correction are, respectively, −6.76 eV and −6.45 eV with the H-H bond length of 0.75 Å. Thus, the μH(T = 0 K) without and with ZPE-correction are −3.38 eV and −3.255 eV. As to the μH(T ≠ 0 K, P), it rests with the temperature and H2 gas pressure. Experimentally, to study the retention of H isotopes under low energy high flux plasma irradiation at the different temperature in W [Citation49], the background and working procedure of H2 gas is less than or close to one atmosphere pressure (i.e. P = ∼105 Pa) around the sample holder. In order to well compare with experiments, here we will directly use the temperature-dependent μH(T ≠ 0 K, P1atm), which is given as μH(T ≠ 0 K, P1atm) = −1.75 kTln (T/7.55) provided by Ji et al. in the previous study [Citation50] because this result has been derived in detail by Sugimoto and Fukai [Citation47]. Thus, according to EquationEquation (4)
(4)
(4) , the total H chemical potential at one atmosphere pressure can be written as
(5)
(5)
At the low concentration case, the equilibrium concentration of H dissolving in W and W-based alloys can be established according to the Sievert's law and it is given as
(6)
(6)
where P and P0 represent the background and reference pressures, respectively. The reference pressure is generally chosen as one atmosphere pressure. The formation entropy ΔS is equal to −4.7k for H in W and W-base alloys [Citation51].
2.3. Computations of H diffusion in W and W-alloy
According to Arrhenius diffusion equation, the diffusivity of an H atom random migrating between two interstitial sites in metal [Citation52] is given by
(7)
(7)
where D0 is diffusion pre-exponential factor, GmH is the H-migrating energy barrier and it is the difference between the energies of H at the transition state (GtsH) and the initial state (GisH), i.e.
(8)
(8)
Wert and Zener’ theory [Citation53] gives that D0 is defined as for the impurity atom along the diffusion route in a cubic metal, and here n, λ and m are the number of the nearest neighboring interstitial position, the jumping length and the mass of H atom, respectively.
According to the transition state theory [Citation54], the diffusivity can be also given as
(9)
(9)
EquationEquation (9)(9)
(9) is a general expression for the impurity diffusion in solid, where ZtsH and ZisH are the total partition functions of the transition state and the initial state, respectively. Also, this formula can be solved for the diffusivity employing either classical or quantum mechanical solution for the vibration partition function. Employing the quantum mechanical solution of the vibration partition function, within the harmonic oscillation approximation, the diffusion rate is written as.
(10)
(10)
where ω+i and ωi are, respectively, two real normal vibration modes for the transition state and the initial state. N represents the number of the vibrating atoms. Note that along the reaction coordinate, the potential energy exhibits a negative curvature so as to yield an imaginary vibration mode (ω*) at the transition state (saddle point). Therefore, there is one real normal mode less at the transition state than at the initial state. The climbing image nudged elastic band (CI-NEB) method [Citation55] was employed to determine minimum energy migration route for the diffusion of H at the interstitial position.
To specifically illustrate the H vibration modes in W and W-alloys, we evaluate the vibration modes of H at the tetrahedral site (TS) and the octahedral site (OS) as well as transition state in the harmonic approximation. According to the quasi-harmonic approximation method of the lattice oscillation, there are 3 N vibration frequencies mentioned in EquationEquation (3)(3)
(3) in a supercell system including N atoms so that each atom exhibits three vibration frequencies. The vibration frequencies of H at ground state are listed in . In the TS, H has three real vibration modes with two degenerate frequencies and one smaller frequency. This indicates that the TS for H is one stable ground state. For the transition state of H diffusion, there are two real vibration frequencies (normal vibration modes) and one imaginary frequency (imaginary vibration mode) in reference to the negative curvature of the saddle point in the direction of the reaction path, based on CI-NEB method [Citation55]. In the OS, however, H has one real vibration frequency and two imaginary frequencies, meaning that the OS for H is only one saddle-point position (unstable state).
Table 1. The vibration frequencies (ω in unit of THz) of H at the TS (ωTS) and OS (ωOS) as well as transition states (ωTran) in W and W-based alloy at 0 K
3. Results and discussion
3.1. Establishment of the temperature-dependent lattice parameter
For the temperature effect in this work, we consider two-part contributions, i.e. the thermal expansion of lattice and vibration free energies of the system. The thermal expansion of lattice rests with the increase of the temperature-dependent lattice parameter. Previously, there were many studies on the thermal expansion behaviors of different structural metals by experimental and theoretical methods. Recently, Lu et al. [Citation56] have given the thermal expansion coefficients of body center cubic (bcc) W using the PARROT module [Citation57], and the obtained data are consistent with the reported experimental results. Although the 0-K lattice parameter of 3.16 Å obtained by Lu et al. is in agreement with that (3.17 Å) calculated in this work, these temperature-dependent lattice parameters given by Lu et al. cannot be directly applicable for our current DFT calculations because of the small difference between Lu et al. and this work. However, we may use the similar analytical method proposed by Lu et al. to correlate the temperature with the lattice parameter. Our temperature-dependent lattice parameter (aT) can be calculated to use in applying our DFT data by multiplying the ratio a'T/a'0 given by Lu et al. with the current optimized 0-K lattice parameter (a0), i.e. aT = a0 · (a'T/a'0). Employing the above method, each will be obtained from first-principles calculations as the ground-state energy. According to EquationEquation (3)
(3)
(3) , Fvib(T) is related to the vibration modes (ωi) of the system at a given temperature. The vibration modes and aT are one-to-one relationship, thus the vibration modes of the system will be re-calculated at each given aT. After establishing the
and Fvib(T), the formation energy and diffusion energy barrier of H at a given temperature in W and W-based alloys can be calculated by substituting the corresponding ‘
’ into EquationEquations (1)
(1)
(1) and (Equation8
(8)
(8) ). Finally, we can get how the formation energies and energy barriers of H in W and W-based alloys alter with the increasing temperature.
To investigate the effect of thermal expansion on H formation and diffusion in W-based alloy, we first calculate the temperature-dependent lattice parameter of W-based alloy. Here, we need point out that only one substitute Cr or V atom is added into W supercell system to form a much dilute W0.9922Cr0.0078 or W0.9922V0.0078 alloy. Therefore, we believe that one AE Cr or V will have little influence on the total W supercell changing with the temperature so that we can obtain the temperature-dependent lattice parameters of W as those of the dilute W–Cr or W–V alloy. As mentioned above, the method was reported by Lu et al. [Citation56] who characterized the molar volumes of W, which is given as
(11)
(11)
where VT and V0 are the molar volumes at the temperature T and 0 K, respectively. Considering the ratio (aT/a0) of lattice parameter, EquationEquation (11)
(11)
(11) will further expressed as
(12)
(12)
According to EquationEquation (12)(12)
(12) , we can obtain the temperature-dependent lattice parameters of W and W-based alloys, as listed in .
Table 2. The ratio (aT/a0) of lattice parameter, the lattice parameter (aT) and the lattice expansion strain (%) of W-based alloy at the relevant temperature
3.2. Formation of H in interstitial lattice at ground state in W-based alloys
Due to their large atom size, the AEs are generally believed to substitute W atoms in the lattice site. This has been really confirmed by our current calculations, where we find that the formation energies of the AEs Cr and V in substitute positions in W are 0.43 eV and −0.51 eV, respectively, consistent with recent results [Citation58].
For H atom, there are two possible interstitial positions in perfect W, i.e. TS and OS. The preferable site for one H atom is the TS, which has been confirmed by our previous work [Citation11]. However, it is important to check whether the site preference of H atom alters or not when H atom is located at the vicinity of the substitute Cr or V element in W; due to that this is the cornerstone of the following study. Around the substitute Cr/V in W atomic structure plane supercell (top view from the [001] direction), the different neighbor TS and OS are shown in (a) and (b), respectively. Here, we point out that the AE Cr/V is exactly placed into the substitute center of 128-atom supercell system since the supercell is made up of 4 × 4 × 4 unit cells, as given in Section 2.2. Accordingly, the distances of the first nearest neighbor (1nn), 2nn, 3nn, 4nn TS (OS) are (a/2),
(
),
(
), and
(3a/2), respectively. We first calculate the formation energies of H initially placed in the 1nn TS/OS of the Cr/V. After structural optimization, we found that the position of the H initially occupying the OS remains nearly unchanged around the Cr/V, whereas the H atom initially placed in the TS has been off the perfect TS close to an OS in the vicinity of Cr/V, and the deviation distance is about 0.23 Å from the original TS. However, it can be noted that the deviation distance is not very large, and thus we still call this site as the quasi-TS in dilute W–Cr and W–V alloys. We further find that the formation energies of H in the quasi-TS are still lower than those in the OS in both W–Cr and W–V alloys. This result fully demonstrates that the H atom is still preferable to occupy the TS over the OS in the vicinity of both AE Cr/V in W. Additionally, we calculated the formation energies of H atom located at the 2nn, 3nn and 4nn TS and OS surrounding the AE Cr/V. The calculated results indicate that the TS is always more energetically favorable than the OS in dilute W–Cr or W–V alloy. Thus, in the following analysis we will only give the corresponding discussions on the situation of H at the 1nn TS in dilute W–Cr and W–V alloys. Our choice of choosing 1nn TS as a research object is mainly because our purpose of present study is to investigate the effect of the AE on the H dissolution in W.
Figure 1. (Top view from the [001] direction) The different neighbor TS (a) and OS (b) around the substitute Cr/V (gray ball) in W (red ball) atomic structure plane supercell. The distances of 1nn, 2nn, 3nn, 4nn TS (OS) are (a/2),
(
),
(
), and
(3a/2), respectively. The TS and OS are denoted by triangle and circle, respectively.
![Figure 1. (Top view from the [001] direction) The different neighbor TS (a) and OS (b) around the substitute Cr/V (gray ball) in W (red ball) atomic structure plane supercell. The distances of 1nn, 2nn, 3nn, 4nn TS (OS) are 5a/4(a/2), 13a/4(2a/2), 29a/4(5a/2), and37a/4(3a/2), respectively. The TS and OS are denoted by triangle and circle, respectively.](/cms/asset/dd14d9c0-202f-4823-9d18-798fddd3e59d/tnst_a_1383210_f0001_oc.jpg)
3.3. Finite-temperature total H formation energy in reference to the temperature-dependent H chemical potential
Employing EquationEquation (1)(1)
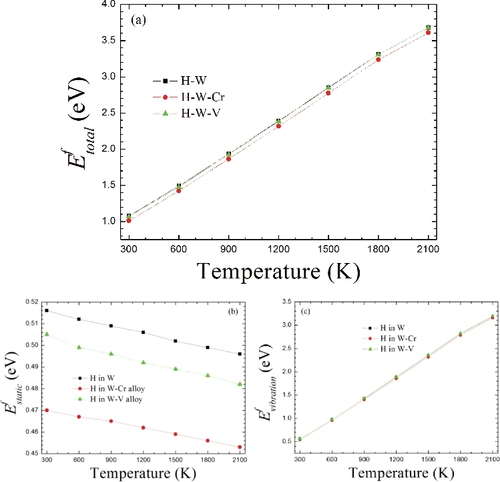
(1) , we calculate the formation energies of H in the TS with the increasing temperature in W–Cr and W–V alloys. For comparison, we also show the corresponding results in pure W, as plotted in . The H formation energies in the TS increase with the increasing temperature in W and W-based alloys ((a)). This demonstrates that the heat of solution of H dissolving in W and W-based alloys will increase with the increase of temperature. In other words, the dissolution of H becomes more and more difficult with the increasing temperature. The definition of formation energy states that the negative formation energy should be an exothermic process, while the positive formation energy represents an endothermic process. According to the current results from (a), one sees that all the formation energies are positive over the whole temperature range from 300 to 2100 K, meaning that the solution process of H in W and W-based alloys is always endothermic with the considered temperature range. Furthermore, we note that with the increasing temperature, the H formation energy is always lower in dilute W–Cr alloy than in dilute W–V alloy as well as pure W, and the energy difference between H in W–Cr and W–V alloys is close to 0.04 eV. This means that the dissolution of H becomes easier in the vicinity of Cr than in the vicinity of V in W. While the formation energy difference of H in W–V alloy and pure W is very small and only ∼0.015 eV, suggesting that the presence of AE V can hardly enhance the H dissolution over the whole temperature regime. Therefore, the appearance of AE Cr in W should be favorable for the H dissolution, while V has little effect on the dissolution of H in W.
Figure 2. The formation energies of H at the TS with the increasing temperature in W and W-based alloys. (a) The total formation energy in reference to the total H chemical potential μH(T, P1atm) = μH(T = 0 K) + μH(T ≠ 0 K, P1atm). (b) The corresponding static formation energy in reference to the static H chemical potential μH(T = 0 K) based on EquationEquation (13)(13)
(13) and it is related to the lattice expansion. (c) Phonon vibration formation energy referring to the temperature-dependent H chemical potential μH(T ≠ 0 K, P1atm) based on EquationEquation (14)
(14)
(14) .
In order to understand specifically the effect of temperature on the H formation energy in dilute W-based alloys, we decompose the total formation energy into two part energies, i.e. the thermal expansion energy and phonon vibration energy. The thermal expansion contribution is the static energy from the DFT calculation for each strained lattice constant aT at each given temperature, then the H formation energy GfH(strain) can be given as.
(13)
(13)
where
and
, respectively, denote the DFT static energies of H–W-alloy and pure W-alloy system for each strained lattice constant aT at a given temperature. While the H formation energy of phonon vibration contribution can be written as
(14)
(14)
where
and
, respectively, are phonon vibration energies of H–W-alloy and pure W-alloy system for each strained lattice constant aT at a given temperature. μH(T ≠ 0 K, P1atm) is equal to −1.75 kTln(T/7.55). Actually, here the H chemical potential has been divided into two parts, i.e. the static μH(T = 0 K) and the temperature-dependent μH(T ≠ 0 K, P1atm).
(b) shows the changing curves of the H formation energy from the thermal expansion contribution with the increasing temperature in W and W-based alloys, respectively. For all the cases, the H formation energy decreases in a close-linear form with the increasing temperature (strain). This demonstrates that the thermal expansion (note that here the thermal expansion corresponds to the isotropic tensile strain) can reduce the H formation energy in W and W-based alloys. The reduced energies from 300 to 2100 K is about 0.02 eV for H in both W and W-based alloys. Also, it can be clearly seen that with the increasing temperature, the H formation energy is always lower in the dilute W–Cr alloy than in the dilute W–V alloy as well as pure W. The energy difference of the thermal expansion contribution between W–Cr and W–V alloy is always ∼0.04 eV, which is not dependent of the temperature. Such a behavior could be understood according to the linear elasticity theory provided by Zhou et al. [Citation16]. According to their study, the H formation energy under tensile strain (thermal expansion) can be obtained as Efϵ = Eϵ = 0f + (σ[100]ϵ[100] + σ[010]ϵ[010] + σ[001]ϵ[001])Vϵ = 0, where Efϵ = 0 is the H formation energy without strain, σ[mln ] is the thermal expansion-induced lattice stress at each crystal-direction in the equilibrium system, ϵ is the strain, and Vϵ = 0 is the volume of the system without strain (here, the system is W/W-based alloy supercell) at equilibrium state. For the present thermal expansion (isotropic strain), ϵ[100]=ϵ[010] = ϵ[001] due to that W and W-based alloys belong to the cubic crystal system, while σ[100] + σ[010] + σ[001] can be approximately equal to (
is the average stress and it is given as
); thus, the H formation energy can be further expressed as
. It can be seen that there is a linear dependence of H formation energy Efϵ on strain ϵ with a slope
. Due to that the average stress
is compressive (negative by convention when the lattice is stretched in our calculations), the H formation energy negatively changes in a close-linear form with the increasing tensile strain in both W and W-based alloys. Therefore, we can predict that the H formation energy from the lattice thermal expansion contribution in W and W-based alloys shows nearly the same behavior under the isotropic strain and therefore to the increase of temperature.
Different from the lattice thermal expansion contribution, the H formation energy from phonon vibration contribution shows an obvious temperature-dependent behavior in W and W-based alloys, as shown in (c). Based on EquationEquation (14)(14)
(14) , the energy reference point is not the H chemical potential μH(T = 0 K), but the temperature-dependent μH(T ≠ 0 K, P1atm) = −1.75 kTln (T/7.55). It is very clear to see that the H formation energies in W and W-based alloys increase in quasi-linear form with the increasing temperature, although the phonon vibration energy is relevant with the term of the ∼exp ( − ℏωi/kT) form given in EquationEquation (3)
(3)
(3) . This is because the absolute value of phonon vibration energy is far lower than that of H chemical potential μH(T ≠ 0 K, P1atm) at the corresponding temperature. For example, the phonon vibration energies of H at T = 1500 K in W, W–Cr alloy and W–V alloy are only 0.043 eV, 0.009 eV and 0.053 eV, respectively, while the μH(T = 1500 K, P1atm) is close to −1.19 eV. Therefore, according to EquationEquation (14)
(14)
(14) , the temperature-dependent H chemical potential directly dominates the temperature-dependent H formation energy so that the energy-temperature curve exhibits a close-linear changing trend ((c)). Here, it should be pointed out that we have checked vibration entropy that does not affect the temperature-dependent H formation energy in both W and W-based alloy, since the contribution of vibration entropy is nearly negligible for the H formation energy with the rising temperature. For instance, we have estimated the free energy contribution from the vibration entropy of H at T = 900 and 1800 K in W, which are about two orders of magnitude smaller than the H formation energy and thus can be neglected. On the other hand, unlike the situation of the thermal expansion energy contribution, the vibration energy contribution in both W and W-based alloys exhibits nearly the same temperature-dependent behavior (see (c)). The energy differences among them can be negligible with the increasing temperature.
3.4. Finite-temperature total H formation energy referring to the static H chemical potential
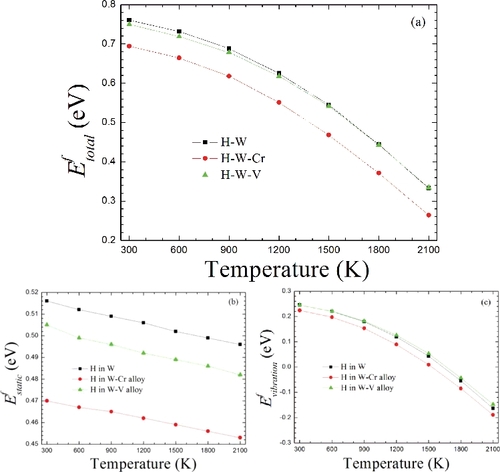
The above results demonstrate that the total formation energies (two parts: the lattice thermal expansion and phonon vibration) of H increase with the increasing temperature at one atmosphere pressure in W and W-based alloys. It is interesting to take into account whether the changing trend of the total H formation energy with the temperature alters or not when the static H chemical potential μH(T = 0 K) is only used as the energy reference point. To answer this question, we have calculated the total H formation energy merely referring to μH(T = 0 K) with the rising temperature. Note that although such an analysis might be not in accordance with conventional physical logic, our purpose is here to compare with the results of the above part (Section 3.3). Surprisingly, there will be an opposite conclusion on the total H formation energy with the increasing temperature in both W and W-based alloys, i.e. the total H formation energy decreases with the increasing temperature, as shown in (a). Similar to the above analysis method, the total H formation energy can be also decomposed into two part contributions, i.e. thermal expansion (GfH(strain)) and phonon vibration (GfH(vibration)), which are, respectively, given as.
(15)
(15)
and
(16)
(16)
Figure 3. The formation energies of H at the TS with the increase of temperature in W and W-based alloys. (a) Here, the total formation energy only referring to the static H chemical potential μH(T = 0 K). (b) The corresponding static formation energy in reference to the static H chemical potential μH(T = 0 K) based on EquationEquation (15)(15)
(15) and it is related to the lattice expansion. (c) Phonon vibration formation energy. Here, it does not refer to any H chemical potential since it is only the difference between H–W-alloy supercell and pure W-alloy supercell, according to EquationEquation (16)
(16)
(16) .
EquationEquation (15)(15)
(15) is equal to EquationEquation (13)
(13)
(13) , whereas EquationEquation (16)
(16)
(16) denotes phonon vibration energy difference between H–W-alloy supercell and pure W-alloy supercell, i.e. without referring to any chemical potential.
According to EquationEquations (15)(15)
(15) and (Equation16
(16)
(16) ), we recalculate the formation energy of H at the TS with the increase of temperature in W and W-based alloys. (b) gives the formation energy of H in reference to the static H chemical potential μH(T = 0 K)with the increasing temperature. It is expected that the H formation energy in both W and W-based alloys from thermal expansion contribution displays the same changing trend as the above results, due to that EquationEquation (15)
(15)
(15) is equal to EquationEquation (13)
(13)
(13) . Employing EquationEquation (16)
(16)
(16) , we further analyze the H formation energy from phonon vibration energy contribution. As clearly seen from (c), due to that the vibrational Helmholtz free energy involves the exponential term (∼exp ( − ℏωi/kT)), the H vibration formation energies in W and W-based alloys exponentially decrease with the increasing temperature, and all the values reduce about 0.40 eV when the temperature changes from 300 to 2100 K. In order to demonstrate the validity of these results, we need to discuss the anharmonic effect on the H formation energy from vibration contribution when the temperature alters from 300 to 2100 K. This is because phonon frequencies are the volume dependence. Physically, Grüneisen constants (γ) can effectively describe the vibration frequency (ω) with the change of lattice volume (V), i.e.
[Citation59], this formula clearly indicates that the vibration frequency decreases with the increase of the lattice volume. Therefore, an expansion of the lattice will result in the softening of vibration modes of all the atoms in material system. Taking H atom in W as an example, the calculated vibration modes of H at the TS have two degenerate frequencies and one smaller frequency, respectively, as listed in . The first degenerate frequency can be found to shift from 46.12 to 43.83 THz if the volume is expanded by 3.232% (corresponding to the temperature increasing from 0 to 2100 K), and then the H vibration formation energy GfH(vibration) = ΔFaTvib(T) can increase slightly, ∼0.02 eV, when the vibration frequencies of the system with the lattice parameter of 3.205 Å (2100 K) are employed instead of the system with the lattice parameter of 3.171 Å (0 K). Therefore, according to our calculations, it is reasonable to speculate a relatively weak anharmonic effect for H in W even at high temperature 2100 K. Moreover, to some extent, it is also reasonable to employ the vibration frequencies of the system with the optimized lattice parameter at all temperatures (see ). Similar results have been found for impurity carbon in Pd and Pd-based alloys [Citation60]. At the same time, it can be noted that the H vibration formation energy in W and W–V alloy exhibits nearly the same the value at each given temperature, while the H vibration formation energy in W–Cr alloy is lower than those in W and W–V alloy. The difference of the vibration formation energy of H in W–Cr alloy and W–V alloy (as well as pure W) is always ∼0.03 eV, independent of the temperature. This should be understood by the different vibration modes of H in W–Cr and W–V alloys, as listed in .
In the above discussions, we gave the change of the vibration formation energy of H at the TS with the temperature in W or W-based alloy. However, a crucial question arises of whether there is any possibility to see that the OS-H becomes more stable than the TS-H with the temperature, especially at high temperature, since the stability of H at the interstitial site can be determined by some kind of balance between amplitude of H vibration and thermal expansion of metal matrix. Therefore, we examined this situation and found that the TS for H atom is always more stable than the OS over the whole considered temperature range in W and W-based alloys. For example, using EquationEquations (15)(15)
(15) and (Equation16
(16)
(16) ), the total H formation energy at the OS in W/W–Cr/W–V are calculated to be 0.88/0.81/0.88 eV and 0.83/0.76/0.83 eV at temperatures T = 1800 and 2100 K, respectively. As potted in (a), at temperatures T = 1800 and 2100 K, the total formation energy of H at the TS in W/W–Cr/W–V are 0.44/0.37/0.44 eV and 0.33/0.26/0.33 eV, respectively, which are significantly lower than those at the OS. Therefore, the TS-H is invariably more stable than the OS-H over the whole temperature region 300–2100 K in both W and W-based alloy.
3.5. Solubility of H in W and W-based alloys
The above study employs two different calculated methods to determine the formation energy of H at the TS in W and W-based alloys, correspondingly resulting in different changing trends of the H formation energy with the temperature. This should be attributed to two different choices of H chemical potential. If we choose the static H chemical potential μH(T = 0 K)as the energy reference point, the H formation energy will gradually reduce with the increase of temperature. If the energy reference point is chosen as the temperature-dependent H chemical potential μH(T, P1atm), the H formation energy will increase significantly with the increasing temperature. Objectively speaking, the temperature-dependent H formation energy in reference to the temperature-dependent H chemical potential should be more in line with the actual case. In other words, the H formation energy referring to μH(T, P1atm) should increase with the temperature in the actual materials. Physically, it is well known that the total energy of the whole material system will increase with the increasing temperature because the electrons will produce the violent motion with the temperature [Citation61,Citation62]. If H or other impurity wants to live in a higher energy system, it will have to get a much higher energy from the external environment to overcome such a higher energy system. This will directly result in an increase of the H formation energy in the higher energy material system. In view of this, in the following solubility calculations, we only take into account the H formation energy obtained by referring to the temperature-dependent H chemical potential μH(T, P1atm).
Using EquationEquation (6)(6)
(6) , we calculate the H solubility as a function of the reciprocal of temperature from 300 to 2100 K at one atmosphere pressure in W and W-based alloys, as shown in . Interstitial H concentrations in both W and W-based alloys increase with the temperature as expected. Generally, the lower the H formation energy is, the higher the dissolving concentration of H in the interstitial site is in material. According to the above calculation, the H formation energy in W–Cr alloy is always lower than that in W–V alloy as well as pure W over the whole temperature range. Thus, interstitial H concentration in W–Cr alloy is always higher than that in W–V alloy as well as pure W, as plotted in . The agreement with experiment is an important criterion to validate the theoretical calculations. In pure W, the typical H solubility equation at one atmosphere pressure is experimentally given as S(H/W) = 9.30 × 10−3exp(−1.04 eV/kT) at temperatures from 1100 to 2400 K, which was reported by Frauenfelder [Citation63]. This experimental data were usually believed to be the most reliable data and have been widely applicable for many experimental and theoretical researches. It can be noted from that at temperature regime from 1200 to 2100 K, our predicted H solubility exhibits a significant deviation from the experimental data and is about two orders of magnitude lower than Frauenfelder's data. This suggests that in W and W-based alloys, the H solubility should be strongly influenced by some factors, which are not taken into account in the current study. First, it is well known that defects such as vacancies and dislocations as well as stacking faults must appear in material. Both vacancies and stacking faults have been demonstrated to serve as the capturing center of H atoms in many transition metals [Citation11,Citation17,Citation64]. Moreover, these captured H atoms can significantly weaken and break the intrinsic metallic bonds in the vicinity of vacancy and stacking fault, which in turn leads to further accumulation of more H atoms in the vicinity of vacancies and stacking faults. For example, Ding et al. [Citation64] predicted a great accommodating ability of the stacking fault for H atoms to be ∼3.40 × 1015 cm−2, agreeing with the experimentally observed H bubble in W-based fusion material. Next, high energy neutron irradiated W as well as W-based alloys leads inevitably to a local strain so as to cause the lattice expansion or compression. Accordingly, strain may change the solubility of H in W and W-based alloys. Recently, Zhou et al. [Citation16] discovered that the H solubility can be enhanced by anisotropic strain in some transition metals. Their results demonstrated a cascading effect of H blistering formation in transition metals, i.e. the H dissolution can result in H blistering formation that causes the local strain that by turns promotes H solubility to further enhance blistering growth. In addition, some transition solutes may still remain in the manufacturing process of W material, accordingly they can strongly interact with H to further affect the H dissolution in W. For instance, Kong et al. [Citation33] carried out first-principles ground-state (0 K) simulations to study the interaction of transition solutes with H in the interstitial lattice as well as vacancy in W and found that most interactions between interstitial H and solutes are attractive, which is consistent with our current result that the temperature-dependent H formation energy can be reduced due to the presence of Cr/V in W, as shown in (a). This result also means that some transition solutes may capture multiple interstitial H atoms to enhance the H solubility in W. Moreover, Kong et al. [Citation33] also demonstrated that there are the large positive binding energies among transition solute, H and defect-vacancy, which further suggests that the strong interaction between solute and defect vacancy can significantly affect the H solubility in W. Thus, these above factors can fully interpret why the experimental data of H solubility is higher than the theoretical results from current calculations in defect-free W.
Figure 4. Solubility of H as a function of the reciprocal of temperature at one atmosphere pressure in W and W-based alloys. The experimental solubility of H in W is given from Frauenfelder [Citation63].
![Figure 4. Solubility of H as a function of the reciprocal of temperature at one atmosphere pressure in W and W-based alloys. The experimental solubility of H in W is given from Frauenfelder [Citation63].](/cms/asset/fd126c17-7cb8-484d-ab19-03f8a16ad44b/tnst_a_1383210_f0004_oc.jpg)
3.6. Finite-temperature H diffusion behaviors in W and W-based alloys
Now, we investigate the diffusion behaviors of H at finite temperature in W and W-based alloys. In pure W, the TS for H is the most stable location. Thus, there are two possible migrating routes. In the first, H atom moves to one 1nn TS passing through a ‘saddle-point’ position. Then we name this route as t→t route. In the second path, H atom migrates to a 2nn TS, passing through an OS, correspondingly we call this route as t→o→t route. The detailed definition of migrating route can refer to CitationRef. 4. For the t→t route, it is well known that the transition state of H is located in a position which is off-middle-position between two 1nn TSs close to an OS. As to the t→o→t route, the OS is just the saddle point so that the diffusion energy barrier is the energy difference of H at the TS and OS. At the ground state, the favorable diffusion path for H atom in W is the t→t route, where the migrating energy barrier is about 0.20 eV, less than the value for the t→o→t route, ∼0.38 eV. Thus, the optimal diffusion path for H atom in W is the t→t route. This has been indeed confirmed by our or other previous works [Citation4,Citation11,Citation13,Citation17]. It is important to take into account whether the optimal diffusion path of H atom alters or not when there is a substitute AE atom nearby in W. To solve this problem, we examine the H energy barrier in the vicinity of Cr or V along the t→t and t→o→t routes, respectively. shows the different neighbor (1nn, 2nn and 3nn) t→t and t→o→t diffusion routes around the substitute Cr/V in W atomic structure plane supercell (top view from the [001] direction). The calculated results indicate that around the AE Cr and V, the H energy barriers for the 1nn t→t route are about 0.10 and 0.15 eV, respectively, less than the values for the 1nn t→o→t route, ∼0.27 and ∼0.32 eV, respectively. We further investigated the energy barriers of H along the 2nn and 3nn t→t and t→o→t routes of the AE Cr or V, and found that the t→t route for H migration is still more favorable than the t→o→t route. Moreover, the diffusion routes are much far away from the AE Cr or V, the H-migrating energy barriers are much close to those in pure W. Thus, similar to the above H dissolution analysis, here we still discuss only the 1nn t→t and t→o→t routes of the AE Cr or V so that we can effectively obtain the influence of the AE on the H diffusion in W.
Figure 5. (Top view from the [001] direction) The different neighbor (1nn, 2nn and 3nn) t→t and t→o→t diffusion routes around the substitute Cr/V (gray ball) in W (red ball) atomic structure plane supercell. The red and black arrows represent t→t and t→o→t routes, respectively. The TS and OS are denoted by triangle and circle, respectively.
![Figure 5. (Top view from the [001] direction) The different neighbor (1nn, 2nn and 3nn) t→t and t→o→t diffusion routes around the substitute Cr/V (gray ball) in W (red ball) atomic structure plane supercell. The red and black arrows represent t→t and t→o→t routes, respectively. The TS and OS are denoted by triangle and circle, respectively.](/cms/asset/dd8d4902-8406-46c2-bec1-c6769e7a2afc/tnst_a_1383210_f0005_oc.jpg)
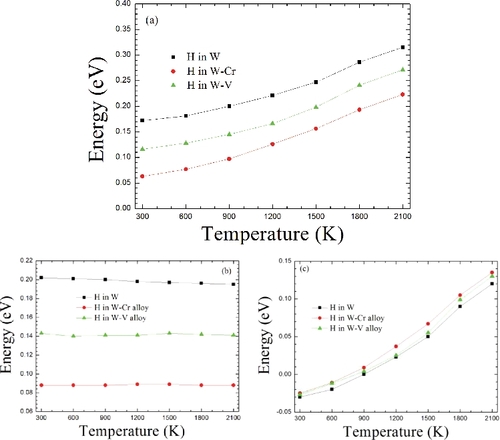
(a) gives the predicted temperature-dependent energy barriers of H along the t→t route in W and W-based alloys. It is important to note that the energy barrier of H migrating along the t→t route in W increases with the increasing temperature, and the value alters from 0.17 to 0.32 eV with the temperature from 300 to 2100 K, consistent with recent calculated results [Citation17]. With the AE additions, we find that the H-migrating energy barriers in both W–Cr and W–V alloys exhibit the same changing trend with the temperature as in pure W. The H energy barriers increase from 0.06 to 0.22 eV and from 0.12 to 0.27 eV when the temperature changes from 300 to 2100 K in W–Cr and W–V alloys, respectively. These results mean that more and more energy should be required for H migrating between two nearest neighbor TSs in W and W-based alloys. On the other hand, we can note that the alloying Cr and V additions have no effect on the changing trend of H-migrating energy barrier with the temperature, but they have large consequences on the absolute values of H-migrating energy barrier at each given temperature. As shown in (a), the presence of V can decrease the H energy barrier to some extent, while the existence of Cr can pronouncedly reduce the H energy barrier. At each given temperature point from 300 to 2100 K, the absolute values can be reduced by ∼0.05 and 0.10 eV in W–V and W–Cr alloys, respectively. Therefore, we deduce that the AE Cr or V addition can be more favorable for H diffusion in W.
Figure 6. Along the t→t diffusion route, the H-migrating energy barriers with the temperature in W and W-based alloys. (a) The total energy barriers. (b) The energy barriers from the lattice expansion contribution. (c) The energy barriers from phonon vibration contribution.
To gain some physical insights on why the H energy barriers increase with the increasing temperature in both W and W-based alloys, we also decompose the total energy barrier into two part, i.e. the thermal expansion and phonon vibration contributions. Based on EquationEquation (8)(8)
(8) , the energy barrier from thermal expansion contribution can be written as
, while the energy barrier from phonon vibration contribution is given as GmH(vibration) = GH(vibration)ts − GisH(vibration). Note that the migrating energy barrier is different from the formation energy and it is independent of the H chemical potential, owing to that it is the energy difference between H at two nearest neighbor TSs. As plotted in (b), the H energy barriers from thermal expansion contribution in W, W–Cr alloy and W–V alloy are invariably 0.20, 0.09 and 0.14 eV, respectively, almost independent of the temperature. However, as seen from (c), the H energy barriers from phonon vibration contribution significantly depend on the temperature and increase with the increasing temperature. When the temperature alters from 300 to 2100 K, the H energy barriers in W, W–Cr alloy and W–V alloy increase from −0.03 eV to 0.12 eV, from −0.025 eV to 0.135 eV and from −0.025 eV to 0.13 eV, respectively. These temperature-dependent properties of H diffusion can be explained by a similar analysis to that applicable for the H formation energy above. Also, the obvious relationship between the total migrating energy barrier and phonon vibration energy contribution can clearly demonstrate that phonon vibration energy contribution dominates the increase of total migrating energy barrier of H along the t→t route with the temperature. By a similar validation to that applied above to the H dissolution, we also found a relatively weak anharmonic effect for H energy barrier even at high temperature.
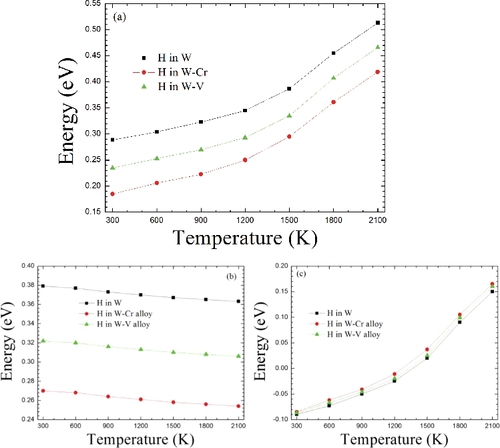
shows the temperature-dependent energy barriers of H along the t→o→t route in W and W-based alloys. Similar to the properties of H along the t→t route, the total energy barrier ((a)) and phonon vibration energy barrier ((c)) of H also increase with the rising temperature, while the H energy barrier from thermal expansion contribution ((b)) hardly depends on the temperature and almost remains constant. However, as observed from (a) and (a) in both W and W-based alloys, the total energy barriers of t→t route are always lower than those of t→o→t route. Therefore, we can infer that the diffusion route of H in the interstitial lattice chiefly advances through the t→t route in W and W–Cr /V alloy. In addition, the changes of phonon vibration energy barriers for t→t and t→o→t routes are 0.15 and 0.24 eV, respectively, when the temperature rises from 300 to 2100 K, as shown in (c) and (c). Such an obvious difference is mainly originated from that the transition states of t→t and t→o→t routes exhibit two and one real vibration modes, respectively, and thus the real vibration modes of transition state of t→t route are one more than that of t→o→t route, as listed in . Note that the OS site is exactly the transition state for H diffusing along the t→o→t.
Figure 7. Along the t→o→t diffusion route, the H-migrating energy barriers with the temperature in W and W-based alloys. (a) The total energy barriers. (b) The energy barriers from the lattice expansion contribution. (c) The energy barriers from phonon vibration contribution.
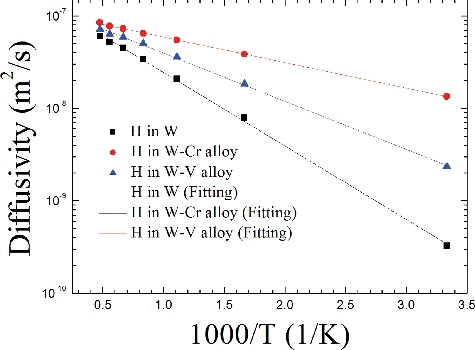
Along the t→t route in W and W-based alloys, the H diffusivity (D) as a function of reciprocal of temperature is presented in . Although the diffusion energy barrier and pre-exponential factor are relevant with temperature, the H diffusivities in both W and W-based alloys still present the nearly quasi-Arrhenius changing trend with the reciprocal of temperature. This suggests that both pre-exponential factor D0 and energy barrier GmH should exhibit the temperature-independent fitted values, which corresponds to exhibit the fitted Arrhenius diffusion equation D = D0exp(−GmH/kT) m2/s, as potted in . Experimentally, many researchers have reported the H diffusivity in W. Among them, Frauenfelder gave the H diffusivity as D = 4.10 × 10−7exp(−0.39/kT) m2/s at temperature range from 1100 to 2400 K [Citation63]. This result was usually believed to be the most credible data, since it was provided at a broad temperature regime and was almost less affected by both surface and defect-capturing influence in W. In the current study, our predicted H diffusivity changes from 3.46 × 10−8 to 6.25 × 10−8 m2/s with the temperature from 1200 to 2100 K, while the experimental H diffusivity alters from 0.95 × 10−8 to 5.02 × 10−8 m2/s at the same temperature regime. Therefore, the order of magnitude (∼10−8 m2/s) of our predicted H diffusivities are in quantitative agreement with experiment. Such a consistency in order of magnitude with experiment for interstitial H diffusion in W suggests that the temperature effect by considering the thermal expansion and phonon vibration contributions is an extremely important factor to accurately interpret experiments and further predict H diffusion behaviors in other metals and metal-alloys. However, for the front coefficients, there are still certain differences between experiment (3.46 and 6.25) and calculation (0.95 and 5.02). This implies that the interstitial H diffusivity in W should be also affected by some factors such as vacancy, dislocation, local strain and possible transition solute, similar to the interstitial H solubility discussed above. Of course, it can be pointed out that the effect of defect distribution on interstitial H diffusivity in metal is still the hardest problem of theoretical estimation. In addition, due to that the experimental data of H diffusivity in W–Cr and W–V alloys are not available up to now, we thus provide a fairly good reference in the future study of interaction between H and W-based alloys.
Figure 8. Along the t→t route, the diffusivity (D) of H as a function of reciprocal of temperature in W and W-based alloys.
4. Conclusions
Based on first-principles total energy and vibrational spectrum calculations, we investigate systematically the finite-temperature H formation and diffusion behaviors in W and W-based alloys. For the temperature effect involving in the H formation energy and diffusion energy barrier, we take into account the contributions of the two parts, i.e. the thermal expansion of lattice and phonon vibration energies. The specific conclusions are as follows.
As to the H formation energy calculations, the tetrahedral interstitial site is only considered in both W and W-based alloys, due to that the tetrahedral interstitial site is always more favorable for H occupying than the octahedral interstitial site. We show that the H formation energies in W and W-based alloys either increase or decrease with the increasing temperature from 300 to 2100 K. There are two cases occurring for the H formation energy with the temperature: one through choosing a static H chemical potential as energy reference point and the other through choosing a temperature-dependent H chemical potential as energy reference point. If we choose the static H chemical potential as the energy reference point, the H formation energy will decrease with the increase of temperature. If the temperature-dependent H chemical potential is chosen as the energy reference point, the H formation energy will increase significantly with the increasing temperature. In both cases, phonon vibration free energy contribution plays a key role in the H formation energy with the temperature, while the thermal expansion energy alteration has little influence on the H formation energy with the temperature. At each given temperature point, the H formation energy is always lower in dilute W–Cr alloy than in dilute W–V alloy as well as pure W, while the H formation energy in dilute W–V alloy is nearly the same as in pure W. These suggest that the presence of AE Cr in W can promote the H dissolution, while the existence of V in W has little effect on the H dissolution. Using the obtained H formation energy referring to temperature-dependent H chemical potential, we further predict the interstitial H solubility at one atmosphere pressure in W, W–Cr alloy and W–V alloy. Our predicted H solubility in W is about two orders of magnitude lower than the reported experimental data at the temperature range from 1200 to 2100 K. Such discrepancy between theoretical results and experimental data is mainly originated from that we do not take into account practical factors such as vacancies, dislocations and local strain, which can effectively capture large amount of H atoms so as to enhance the H solubility in W.
For H diffusion in W and W-based alloys, we only take into account the optimal migrating route t→t route. The H-migrating energy barriers in both W and W–Cr/V alloys significantly increase with the increasing temperature. When the temperature alters from 300 to 2100 K, the H energy barriers in W, W–Cr alloy and W–V alloy increase from 0.17 to 0.32 eV, from 0.06 to 0.22 eV and from 0.12 to 0.27 eV, respectively. This indicates that the H diffusion in both W and W-based alloys becomes more and more difficult with the increasing temperature. Similar to the H formation, phonon vibration free energy contribution plays a crucial role in the H-migrating energy barrier with the temperature, while the thermal expansion energy contribution has little influence on the H-migrating energy barrier with the temperature. At each given temperature point, the alloying Cr or V addition in W has a large consequences on H-migrating energy barrier. The energy barriers can be reduced by ∼0.05 and 0.10 eV in W–Cr and W–V alloys, respectively. Based on the obtained H-migrating energy barriers, the H diffusivities are predicted in both W and W-based alloys. With the temperature from 1200 to 2100 K, the calculated H diffusivity in W alters from 3.46 × 10−8 to 6.25 × 10−8 m2/s. The order of magnitude is in quantitative agreement with the reliable experimental data from 0.95 × 10−8 to 5.02 × 10−8 m2/s at the same temperature regime. Due to that the experimental data of H diffusivity in W–Cr and W–V alloys are not available up to now, thus the current predicted H diffusivity in W–Cr and W–V alloys might provide a fairly good reference in the future study of interaction between H and W-based alloys.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Causey R, Wilson K, Venhaus T, et al. Tritium retention in tungsten exposed to intense fluxes of 100 eV tritons. J Nucl Mater. 1999;266:467–471.
- Shu WM, Wakai E, Yamanishi T. Blister bursting and deuterium bursting release from tungsten exposed to high fluences of high flux and low energy deuterium plasma. Nucl Fusion. 2007;47:201–209.
- Bolt H, Barabash V, Krauss W, et al. Materials for the plasma-facing components of fusion reactors. J Nucl Mater. 2004;329–333:66–73.
- Liu YL, Zhou HB, Zhang Y. Investigating behaviors of H in a W single crystal by first-principles: from solubility to interaction with vacancy. J Alloys Comp. 2011;509:8277–8282.
- Oya Y, Li X, Sato M, et al. Deuterium permeation behavior for damaged tungsten by ion implantation. J Nucl Sci Technol. 2015;53:402–405.
- Kim HI, Kim DH, Lee Y-O. Evaluation of neutron production cross sections of tungsten isotopes for fusion applications. J Nucl Sci Technol. 2014;45:89–92.
- Sato K, Ezato K, Taniguchi M, et al. Proposal of hot-pressed, rod-shaped tungsten armor concept for ITER divertor and its high-heat-flux performances. J Nucl Sci Technol. 2012;42:643–650.
- Meigo S, Takada H, Shigyo N, et al. Measurements of neutron spectra produced from a thick tungsten target bombarded with 0.5- and 1.5-GeV protons. J Nucl Sci Technol. 2014;39:1252–1255.
- Ogawa T, Hasegawa A, Kurishita H, et al. Improvement of surface exfoliation behavior by helium-ion bombardment of a tungsten alloy fabricated by mechanical alloying. J Nucl Sci Technol. 2012;46:717–723.
- Kunieda S, Shigyo N, Ishibashi K. Nuclear data evaluations on zirconium, niobium and tungsten for neutron and proton incidence up to 200 mev. J Nucl Sci Technol. 2012;41:1047–1058.
- Liu YL, Zhang Y, Zhou HB, et al. Vacancy trapping mechanism for hydrogen bubble formation in metal. Phys Rev B. 2009;79:172103–172106.
- Ohsawa K, Goto J, Yamakami M, et al. Configuration and binding energy of multiple hydrogen atoms trapped in monovacancy in bcc transition metals. Phys Rev B. 2012;85:094102–094109.
- Heinola K, Ahlgren T. Diffusion of hydrogen in bcc tungsten studied with first principle calculations. J Appl Phys. 2010;107:113531–113538.
- Heinola K, Ahlgren T, Nordlund K, et al. Hydrogen interaction with point defects in tungsten. Phys Rev B. 2010;82:094102–094106.
- Ohsawa K, Eguchi K, Watanabe H, et al. Trapping of multiple hydrogen atoms in a tungsten monovacancy from first principles. Phys Rev B. 2010;82:184117–184122.
- Zhou HB, Jin S, Zhang Y, et al. Anisotropic strain enhanced hydrogen solubility in bcc metals: the independence on the sign of strain. Phys Rev Lett. 2012;109:135502–135506.
- Kong XS, Wang S, Wu X, et al. First-principles calculations of hydrogen solution and diffusion in tungsten: temperature and defect-trapping effects. Acta Mater. 2015;84:426–435.
- Wampler WR, LaBombard B, Lipschultz B, et al. Molybdenum erosion measurements in Alcator C-Mod. J Nucl Mater. 1999;266–269:217–221.
- Nagata S, Takahiro K. Deuterium retention in tungsten and molybdenum. J Nucl Mater. 2000;283–287:1038–1042.
- Wright GM, Whyte DG, Lipschultz B. Measurement of hydrogenic retention and release in molybdenum with the DIONISOS experiment. J Nucl Mater. 2009;390–391:544–549.
- Hill ML. Diffusion of hydrogen in metals. J Metals. 1960;12:725–734.
- Oates WA, Mclellan RB. The solubility of hydrogen in molybdenum. Scr Metall. 1972;6:349–352.
- Becquart CS, Domain C. An object Kinetic Monte Carlo Simulation of the dynamics of helium and point defects in tungsten. J Nucl Mater. 2009;385:223–227.
- Golubeva AV, Mayer M, Roth J, et al. Deuterium retention in rhenium-doped tungsten. J Nucl Mater. 2007;363:893–897.
- Tyburska-Püschel B, Alimov VK. On the reduction of deuterium retention in damaged Re-doped W. Nucl Fusion. 2013;53:123021–123032.
- Alimov VK, Hatano Y, Sugiyama K, et al. Surface morphology and deuterium retention in tungsten and tungsten-rhenium alloy exposed to low-energy high flux D plasma. J Nucl Mater. 2014;454:136–141.
- Schmid K, Rieger V, Manhard A. Comparison of hydrogen retention in W and W/Ta alloys. J Nucl Mater. 2012;426:247–253.
- Zayachuk Y, Hoen MHJ, van Emmichoven PAZ, et al. Deuterium retention in tungsten and tungsten-tantalum alloys exposed to high-flux deuterium plasmas. Nucl Fusion. 2012;52:103021.
- Zayachuk Y, Hoen MHJ, van Emmichoven PAZ, et al. Surface modification of tungsten and tungsten-tantalum alloys exposed to high-flux deuterium plasma and its impact on deuterium retention. Nucl Fusion. 2013;53:013013.
- Zayachuk Y, Manhard A, Hoen MHJ, et al. Depth profiling of the modification induced by high-flux deuterium plasma in tungsten and tungsten–tantalum alloys. Nucl Fusion. 2014;54:123013.
- Kong XS, You YW, Fang QF, et al. The role of impurity oxygen in hydrogen bubble nucleation in tungsten. J Nucl Mater. 2013;433:357–363.
- Wang S, Kong XS, Wu XB, et al. Effects of nitrogen on hydrogen retention in tungsten: first-principles calculations. J Nucl Mater. 2015;459:143–149.
- Kong XS, Wu X, Liu CS, et al. First-principles calculations of transition metal solute interactions with hydrogen in tungsten. Nucl Fusion. 2016;56:026004–026017.
- Aguirre MV, Martín A, Pastor J Y, et al. Mechanical behavior of W-Y2O3 and W-Ti alloys from 25 °C to 1000 °C. Metall Mater Trans A. 2009;40:2283–2290.
- Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys Rev B. 1993;47:558–561.
- Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54:11169.
- Perdew JP, Chevary JA, Vosko SH, et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys Rev B. 1996;46:6671.
- Perdew JP, Burke K, Ernzerhof M. Perdew, burke, and ernzerhof reply. Phys Rev Lett. 1998;80:891.
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865.
- Blochl PE. Projector augmented-wave method. Phys Rev B. 1994;50:17953.
- Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B. 1999;59:1758.
- Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B. 1976;13:5188.
- Kittel C. Introduction to solid state physics. 7th ed. New York: Wiley; 1996. Chapter 1, Crystal structure; p. 1–17.
- Methfessel M, Paxton AT. High-precision sampling for Brillouin-zone integration in metals. Phys Rev B. 1989;40:3616.
- Reuter K, Scheffler M. Composition, structure, and stability of RuO2 (110) as a function of oxygen pressure. Phys Rev B. 2001;65:035406–035416.
- Van de Walle CG, Neugebauer J. First-principles calculations for defects and impurities: applications to III-nitrides. J Appl Phys. 2004;95:3851–3879.
- Sugimoto H, Fukai Y. Solubility of hydrogen in metals under high hydrogen pressures: thermodynamical calculations. Acta Metall Mater. 1992;40:2327–2336.
- Hemmes H, Driessen A, Griessen R. Thermodynamic properties of hydrogen at pressures up to 1 Mbar and temperatures between 100 and 1000 K. J Phys C. 1986;19:3571–3585.
- Luo G-N, Shu WM, Nishi M. Influence of blistering on deuterium retention in tungsten irradiated by high flux deuterium 10–100 eV plasmas. Fusion Eng Design. 2006;81:957–962.
- Ji M, Wang CZ, Ho KM, et al. Statistical model of defects in Al-H system. Phys Rev B. 2010;81:024105–024108.
- Fukai Y. The metal-hydrogen system. Berlin: Springer; 1993. Chapter 4, Metal-hydrogen system; p. 115.
- Philibert J. Atom movements: diffusion and mass transport in solids. Paris (France): Les editions de Physique; 1991. p. 73.
- Wert C, Zener C. Interstitial atomic diffusion coefficients. Phys Rev. 1949;76:1169–1175.
- Wigner E. Perspective on “the transition state method.” Trans Faraday Soc. 1938;34:29–41.
- Jonsson H, Mills G, Jacobsen KW. Classical and quantum dynamics in condensed phase simulations. London (NJ): World Scientific; 1998. p. 58.
- Lu XG, Selleby M, Sundman B. Assessments of molar volume and thermal expansion for selected bcc, fcc and hcp metallic elements. Comput Coupling Phase Diagrams Thermochem. 2005;29:68–89.
- Jansson B. TRITA-MAC 0234. Stockholm: Royal Institute of Technology; 1984.
- Kong XS, Wu XB, You YW, et al. First-principles calculations of transition metal–solute interactions with point defects in tungsten. Acta Mater. 2014;66:172–183.
- Ashcroft NW, David N. Solid state physics. New York: Cornell University; 1989.
- Ling C, Sholl DS. First-principles evaluation of carbon diffusion in Pd and Pd-based alloys. Phys Rev B. 2009;80:214202.
- Wang Y, Liu ZK, Chen LQ. Thermodynamic properties of Al, Ni, NiAl, and Ni3Al from first-principles calculations. Acta Mater. 2004 52:2665–2671.
- Togo A, Chaput L, Tanaka I, et al. First-principles phonon calculations of thermal expansion in Ti3SiC2,Ti3AlC2, and Ti3GeC2. Phys Rev B. 2010;81:174301.
- Frauenfelder R. Permeation of hydrogen through tungsten and molybdenum. J Chem Phys. 1968;48:3955–3965.
- Ding WY, He HY, Pan BC. Interaction of H with stacking fault in W(111) film: a possible formation mechanism of H bubbles. RSC Adv. 2014;4:7030–7034.