
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Using first-principles calculations, we have performed the detailed researches on La behavior in Mo including structure, thermodynamic stability, interaction with vacancy, and mechanical properties of Mo-La alloy. La prefers to stay at substitution site with the solution energy of 2.86 eV. The <111> mixed dumbbell is more stable than both <100> and <110> mixed dumbbells. The La-La interaction displays attraction and repulsion in short- and long-distance range, respectively. Both 1nn and 2nn shells are most likely to form the La-La clusters since there are relatively larger binding energies, which are 0.43 and 0.33 eV, respectively. The La-vacancy interaction is much local and limited within the 2nn shell range. At 1nn and 2nn shells, the binding energies of La-vacancy are 1.33 and 0.26 eV, respectively. Also, the mechanical properties of Mo1-xLax (0 < x < 0.1) binary alloys have been analyzed in detail. The mechanical quantities of single crystal are the elastic constants, Young’s modulus and shear modulus, while the mechanical quantities of poly-crystal include bulk modulus B, Young’s modulus E, shear modulus G, Poisson’s ratio v, Anisotropy factor A and B/G. These results can provide a much valuable reference for Mo-alloy application in nuclear fusion field.
1. Introduction
Due to its low thermal expansion coefficient, high melting temperature, and thermal conductivity [Citation1–4], molybdenum (Mo) has been widely applied in metallurgy, chemical industry, machinery, and nuclear industry. Especially in nuclear fusion reactor, Mo mirrors will be applicable for the optical diagnostics in order to control the plasma in Tokamak of ITER. Meanwhile, Mo mirrors have to suffer the transient temperature increment [Citation5–8], which can degrade the mechanical properties of Mo. Therefore, it is very necessary to optimize mechanical properties of Mo material, which can help us well operate the optical systems in Tokamak. Accordingly, the alloying of Mo is an effective method to well improve the general performance of Mo. It is well known that many researches have been performed on Mo alloys for several decades [Citation9,Citation10]. For instance, by adding Al, K and Si into perfect Mo, the AKS-Mo has been fabricated and reported to have higher strength, toughness, and re-crystallization temperature than those of perfect Mo [Citation11].
Lanthanum (La) is the typical alloying element in refractory metals, which can form metal-La alloys with excellent corrosion resistance and high-temperature strength characteristics. The formed-Mo alloy by doping La oxide (La2O3) has been attested to be a very good material in industrial production of Mo wire since La2O3 particles can refine the crystalline structure, raise the re-crystallization temperature and improve strength/toughness [Citation12–14]. But, the structural properties as well as production process of Mo-La2O3 alloy can be significantly influenced by the La concentration as well as the doping method. At the same time, La belongs to a kind of rare earth metal and exhibits a much higher price, and thus reducing La concentration can help manufacturer decrease production costs. Additionally, it is very important to explore Mo alloy by doping a small amount of La. For instance, the modern pulse generator (Note that the modern pulse generator is an electrical device that can be used as an internal signal source for circuits. It can be also used as an external signal source for devices by generating constant or sustained signal pulses. The pulse generators are used to transmit signals and measure the amount of signal received by equipment. It can use digital and analog circuits to generate the pulses it provides.) requires much better Mo wire to improve the cutting efficiency. Accordingly, the Mo wire has to possess the ability to be subjected to the high current maximum of 700 A to satisfy the demand of surface finishing, which is dependent on the conductivity.
From the above, it can be seen that the alloying effect of La in Mo is crucial for its application in various fields. To our best knowledge, up to now, the structural and thermodynamic properties of rare earth La in Mo and the effect of La on mechanical properties of Mo have not been studied systematically. Correspondingly, there are few works reporting these relevant researches, especially for the simulation work. This greatly limits the application of Mo-La alloy in the field of the current nuclear fusion Tokamak. In view of this, in this study we have performed systematically first-principles calculations to explore structural and thermodynamic stability of La and its interaction with point defects in Mo as well as the influence of La on mechanical properties of Mo. The point defects are here taken into account containing self-interstitial atoms (SIAs) and vacancy. These research results will not only be quite helpful to understand the interaction between the alloying element La and host Mo material but can also give a guiding opinion for the design of the alloy composition in Mo, which further provide an useful reference and a database for the performance and lifetime evaluation of Mo-La alloy for the optical diagnostics in future nuclear fusion reactor.
2. Methods
We carried out first-principles calculations based on density functional theory (DFT) method by using the pseudopotential plane wave as implemented in the Vienna Ab-initio Simulation Package (VASP) code [Citation15,Citation16]. Similar to recent DFT researches [Citation17–21], the electronic exchange-correlation energy is treated using the generalized gradient approximations (GGA) schemes of Perdew-Burke-Ernzerhof functional [Citation22–24]. The 5s4p4d and 5s4p26d electrons were treated as valence electrons for Mo and La, and the ionic cores were represented by the projector augmented wave potentials [Citation25,Citation26]. The convergence tests have been strictly performed for the total energy with respect to the energy cutoff, and 350 eV is sufficient for the total energy and geometry of Mo supercell with and without the alloying element La. Therefore, the energy cutoff 350 eV is presently employed. During the geometry optimization, we used the 128-atom supercell containing (4 × 4 × 4) unit cells with the lengths of 12.64 Å in the [100], [010], and [001] directions, respectively. For body centered cubic (bcc) Mo single crystal, the calculated equilibrium lattice constant is 3.16 Å, in good agreement with the corresponding experimental value of 3.15 Å [Citation27]. The Brillouin zone of supercell was sampled with the Monkhorst-Pack scheme [Citation28] using a (3 × 3 × 3) k-points mesh. The Methfessel-Paxton [Citation29] smearing method was used to integrate the Brillouin zone and account for partial occupancy of the metals near the Fermi level with a smearing width of 0.1 eV. During the calculations, the supercell size and atomic positions are relaxed to equilibrium. The convergence accuracy of inter-atomic forces and total energy is 10−3 eVÅ−1 and 10−5 eV, respectively.
The vacancy formation energy is given as
where is the total energy of Mo supercell containing one vacancy.
is the total energy of perfect Mo supercell. N denotes 128 Mo atoms, i.e. the number of perfect Mo supercell.
In perfect Mo, the solution energy of La in the interstitial position is written as
where is the total energy of perfect Mo supercell with one interstitial La atom,
is the La chemical potential. The La chemical potential is one atom energy in a hexagonal close packed (hcp) La supercell and it is calculated to be −4.92 eV, in good agreement with previous result of −4.936 from the DFT [Citation30].
The binding energy can evaluate the interaction between La and point defect in Mo. For the interaction between La and point defect as well as two point defects including A and B, the binding energy is written as [Citation31]
where and
denote the total energies of the Mo supercells with A and B, respectively,
represents the total energy of the Mo supercell with both A and B. For the positive binding energy, there is an attractive interaction between A and B; for the negative binding energy, there is an repulsive interaction between A and B.
3. Results and discussion
3.1. Intrinsic point defects in Mo
In order to confirm the accuracy of the present calculations, the formation energies of intrinsic point defects are first investigated in Mo. The formation energy of vacancy is calculated to be 2.59 eV. The reported experimental values are 2.24 eV [Citation32], 2.70–3.24 eV [Citation32] and 3.00 eV [Citation33] using the specific heat, the resistivity, and the positron measurements, respectively. It can be seen that the present calculated result agrees with those of experiments within the error range, as shown in . We also calculated the formation energies of self-interstitial atoms with the following structures: <001>-dumbbell, <110>-dumbbell, <111>-dumbbell, octahedron and tetrahedron. The <111>-dumbbell is the most stable structure, followed by <110>-dumbbell, tetrahedron, <001>-dumbbell and octahedron, which are, respectively, 0.15, 0.96, 1.56, and 1.64 eV higher in energy. Apparently, our calculated data are slightly higher or lower than those of previous simulations and experiments however basically in agreement with each other, as shown in .
Table 1. The formation energy (eV) of vacancy and self-interstitial atoms in Mo.
Recently, different descriptions used to model a point-defect in an elastic continuum are reviewed by Clouet et al. [Citation36]. This elastic correction has been shown to accelerate the convergence of the point-defect formation or migration energies obtained from first-principles calculations in materials such as vacancy in diamond silicon [Citation37], self-interstitial atoms in hcp Zr [Citation37,Citation38] and solute interstitials in bcc iron [Citation39]. Accordingly, the elastic energy should not be negligible for the absolute values of the formation energy of vacancy and self-interstitial atom in the present 128-atom supercell because periodic boundary conditions become more significant. The result of the elastic energy calibration might explain the difference between computation and experiments. However, the elastic energy correction cannot change the order of the relative stability of the formation energies for self-interstitial atoms in Mo. Therefore, the elastic energy does not affect the present conclusion.
3.2. Structural and thermodynamic stability of La atom in Mo
We further investigate structural character and thermodynamic stability of La at three positions including tetrahedron interstitial site (TIS), octahedron interstitial site (OIS), and substitution site (SS) in Mo, as shown in ). presents the solution energies of La at these three sites. The solution energies are 10.57 and 11.19 eV for La at TIS and OIS, respectively. Although the solution energy of TIS-La is lower than that of OIS-La, these two energies are relatively higher and endothermic in Mo. So larger solution energies are mainly originated from that both TIS and OIS cannot provide effective space to hold one La atom. In general, the SS should be the preferred position for the alloying element La (as well as other alloying elements) since the SS possesses the larger space compared to these two interstitial sites. As expected, there is indeed a lower solution energy of 2.86 eV for La atom at SS in comparison with both TIS and OIS, as shown in . This indicates that the dissolution of La at SS is energetically favorable in Mo. For comparison, three mixed dumbbell structures including <100>, <110> and <111> are also explored, as given in . One can find that their solution energies are much higher than that of La at SS, as shown in . Comparing with each other, the <111> mixed dumbbell is more stable than both <100> and <110> mixed dumbbells, which is in agreement with the previous study [Citation34] that Muzyk et al. calculated the solution energies of W-V and W-Ta mixed dumbbells in W and found that the <111> mixed dumbbell is also the most stable configuration.
Figure 1. Atomic structures for La-doped in bcc Mo. (a) substitution site, (b) tetrahedron interstitial site, (c) octahedron interstitial site, (d) <100> mixed dumbbell, (e) <110> mixed dumbbell and (f) <111> mixed dumbbell. The gray and green balls represent Mo and La atoms, respectively. (for interpretation of color in this figure, the reader may refer to the web version of this article).
Figure 2. The solution energies of La atom at the different interstitial sites in Mo.
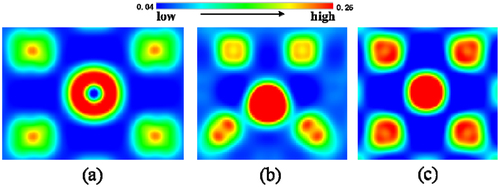
plots the total density of states (TDOS) of La at SS, TIS and OIS, respectively. As can be seen, the TDOS cannot reflect the differences for La at three positions. To accurately describe the interaction of La with nearest neighbor (nn) Mo atoms, we need to give the partial density of states (PDOS) of 1nn Mo with La at different sites, as shown in . It is clear to see that a relatively wide and high peak appears in the low-energy state (from −5.0 to −3.5 eV) when La is located at TIS, while there are no higher peaks when La stays at both OIS and SS. Apparently, there is a strong interaction between TIS-La and 1nn Mo atoms. Valence charge density plays a key role in the analysis of interatomic bonding, as charge accumulation/depletion is a direct indicator of the strengthening/weakening of atomic bond. To further gain the physical insights on the interaction between La and its nn Mo atoms, we show valence charge density distribution for La at three different sites, as shown in . It is clearly seen that the TIS-La interacts strongly with its 1nn Mo atoms since the valence charge density between La and Mo atoms is much higher those of other two cases, suggesting that the TIS-La can form the strong metallic bonds with its 1nn Mo atoms. This corresponds to the higher peak of DOS for the interaction of TIS-La with 1nn Mo atoms mentioned above.
Figure 3. Total density of states (TDOS) of La at TIS, OIS and SS, respectively. The Fermi energy for the respective case is set to be zero.
Figure 4. The partial density of states (PDOS) of 1nn Mo with La at TIS, OIS and SS, respectively. The Fermi energy for the respective case is set to be zero.
Figure 5. The charge density distribution (Unit is e/Å3) for La at different sites in Mo. (a) substitution site, (b) tetrahedron interstitial site and (c) octahedron interstitial site. (For interpretation of color in this figure, the reader may refer to the web version of this article.).
3.3. The binding property between two La atoms in Mo
In order to comprehensively understand the physical behavior of the alloying element La in Mo, it is very necessary to investigate the interplay between two La atoms (La-La pair) which could give an insight for La-La accumulation. Since La atom prefers to occupy the SS, here we put two La atoms into two different SSs as a function of La-La distance, as shown in . According to EquationEquation (3)(3)
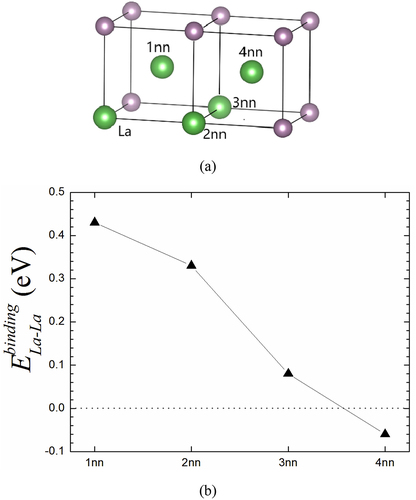
(3) , we have calculated the binding energy of La-La pair from 1nn to 4nn. As given in , the La-La binding energy decreases gradually with the increasing La-La distance. Also, it can be clearly seen that the binding energies of La-La pairs are positive for the first three adjacent shells (from 1nn to 3nn) and then become the negative values for the 4nn shell. This fully suggests that the La-La interaction presents attraction and repulsion in short- and long-distance range, respectively. This also demonstrates that the interaction between two La atoms is much local, constrained largely within the 3nn shell regime. At the first three nn shells, there is a possibility for two La atoms to accumulate to form the small La clusters in Mo-La system. Especially, as shown in , both 1nn and 2nn are most likely to form the La-La clusters since there are relatively larger binding energies, which are 0.43 and 0.33 eV, respectively.
Figure 6. (a) The La-La pair structures for the second La atom at different nearest neighbor (1nn to 4nn) sites relative to the first fixed La atom in Mo. Note that both two La atoms occupy substitution site. (b) The binding energy of La-La pair at substitution sites from 1nn to 5nn in Mo.
3.4. The interaction of La with vacancy in Mo
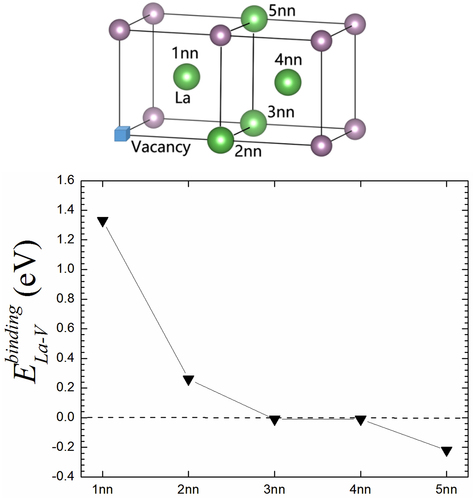
gives the vacancy-La structures for La at different nearest neighbor (1nn to 5nn) SSs relative to the vacancy in Mo system. At the same time, the binding energy between La and vacancy has been calculated, as shown in . One sees that the vacancy-La displays a much strong attractive interaction for La at 1nn site and the binding energy is 1.33 eV. While a weak attractive interaction appears for vacancy-La at 2nn site and the corresponding binding energy is 0.26 eV. This should be a consequence of the fact that the 1nn and 2nn distances are relatively far in bcc Mo structure, and the distance difference between them is close to 15.5%. The similar results have been reported in bcc Fe [Citation35,Citation40]. For fcc Al, the 1nn and 2nn distances have a much large difference with the value of ~ 41%. Significant digits should be same as for bcc metals. Moreover, this value is not for only Al, but for all fcc metals. Previously, Wolverton [Citation41] showed that the interplay between vacancy and the alloying element at 2nn site is much weak and much lower in magnitude than that at 1nn site. Our current results attest the previous conclusion again. Further, the binding energy of vacancy-La at 3nn site is almost equal to zero and vanishes for the large distance, i.e. 4nn and 5nn cases. This suggests that the La-vacancy interaction is much local and limited within the 2nn shell range.
Figure 7. (a) The atomic structures for La-vacancy interaction with La atom at different nearest neighbor (1nn to 5nn) SSs relative to vacancy in Mo system. (b) The binding energy between vacancy and La at SS from 1nn to 5nn of vacancy vicinity.
3.5. Mechanical properties of Mo-La alloy
According to the elastic theory [Citation42,Citation43], there are three independent elastic constants in bcc Mo1-xLax (0<x < 0.1) binary alloy, namely C11, C12 and C44. For cubic crystal system, the elastic constant matrix is given as
To obtain three elastic constants, three sets specific strains (δ) are applied in different directions of bcc Mo1-xLax supercell. According to Hooks’s law, a quadratic function relationship between total energy change () and strain (δ) is established. As given in .
Table 2. The relationship between the total energy change (ΔE) and strain (δ) of cubic crystal system.
For the small strain δ, the total energy E(V,δ) of the strained supercell can be expanded according to the Taylor series of the strain tensor
where E(V0,0) is the total energy of the supercell without strain, V0 is the volume of primary cell without strain, Cij is the elastic constant, and the indices i and j are from 1 ~ 6, is the Voigt index factor,
is the stress tensor element.
3.5.1. Lattice constants
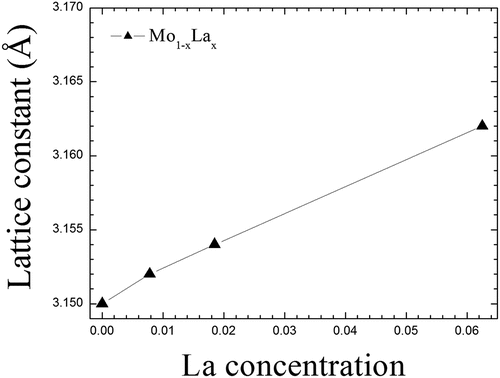
As shown in , the lattice constant alteration of Mo1-xLax (0 < x < 0.1) binary alloy is significantly dependent on the La concentration. There is a substantial linear relationship between La concentration and lattice constant. In metallurgy, Vegard’s law is an approximate empirical rule which holds that a linear relation exists between the crystal lattice parameter of an alloy and the concentrations of the constituent elements. It can be seen that the present Mo1-xLax alloy system indeed follows Vegard’s law. At the same time, the computed lattice constant of Mo1-xLax binary alloy increases with the increasing La content, suggesting that La can enlarge the lattice structure of Mo. Such a changing trend can be well understood from that the atomic radius of La atom is 1.88 Å, obviously larger than that (1.40 Å) of host Mo atom.
Figure 8. The calculated lattice constants for Mo1-xLax (0<x < 0.1) binary alloys as a function of La concentration.
3.5.2. Elastic properties of single crystal Mo1-xLax binary alloy
According to different lattice constant given in , we have calculated the elastic constants as a function of La content. presents the elastic constants Cij of single crystal Mo1-xLax with increasing La content, and the detailed results are also listed in . It can be seen that the effect of La on C11 is much greater than that on both C12 and C44 with the increase of La content. C11 alters from 479.2 GPa of perfect Mo to 442.7 GPa of Mo0.9375La0.0625 alloy with the doping of La and the difference-value between perfect Mo and Mo0.9375La0.0625 alloy is close to 36.5 GPa. This demonstrates that C11 is strongly dependent on the La concentration. Compared to C11 situation, the difference-value for C12 (C44) is 10.8 (12.3) GPa between between perfect Mo and Mo0.9375La0.0625 alloy, implying that both C12 and C44 are weakly dependent on the La content. As shown in , both C11 and C44 display the negative changing trend, i.e. decrease with the increasing La concentration, whereas C12 presents a weak growth tendency with the increase of La amount. The changes in these three mechanical quantities (C11, C12 and C44) can be explained by the above electronic structure calculations (DOS) of Mo-La alloy, as shown in . It can be seen that there is a similar changing trend by comparing the DOSs of SS La atom (nn Mo atom (), i.e. there are varying degrees of overlapping between Mo and La atoms when La is added into SS in the Mo lattice. This fully indicates that the bonding interaction between two atoms appears in Mo-La alloy. Thus, such a bonding interaction changes the elastic constants of pure Mo. In fact, the main characteristic of electronic structures of Mo-La alloy is the hybridization between Mo-d and La-d orbitals.
Figure 9. The calculated elastic constants (Cij in GPa) and tetragonal shear modulus (C’ in GPa) for single crystal Mo1-xLax (0 < x < 0.1) binary alloys.
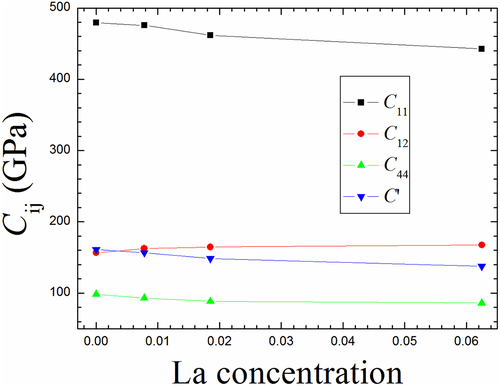
Table 3. The elastic constants (in GPa) and tetragonal shear modulus (C’ in GPa) for single crystal Mo1-xLax (0 < x < 0.1) alloy.
The elastic constant C11 is generally applicable for reflecting the stiffness of metal-alloy materials. As shown in , the elastic constants C11 of all these Mo1-xLax binary alloys are lower than that of perfect Mo, suggesting that the stiffness of perfect Mo can be reduced due to the addition of La. To sum up, the elastic constants C11 of Mo1-xLax binary alloys as well as perfect Mo obey the following order: Mo>Mo0.9922La0.0078>Mo0.9815La0.0185>Mo0.9375La0.0625, indicating that Mo0.9375La0.0625 alloy exhibits the weakest stiffness among these Mo1-xLax binary alloys. This elastic constant C12 is generally used to judging the ability of metal-alloy to resist transverse deformation. As given in , the elastic constants C12 of Mo1-xLax binary alloys is higher than that of perfect Mo, and increases with the increase of La concentration within the current concentration range. This suggests that the doping La in Mo can raise the ability of perfect Mo to resist transverse deformation. The elastic constants C12 of Mo1-xLax binary alloys as well as perfect Mo comply with the following order: Mo<Mo0.9922La0.0078<Mo0.9815La0.0185<Mo0.9375La0.0625, demonstrating that Mo0.9375La0.0625 alloy possesses the strongest ability to resist transverse deformation among these Mo1-xLax binary alloys. Furthermore, the mechanical stability criterion is (C11-C12) > 0, (C11 +2C12) > 0, C11 >0 and C44 >0 for cubic crystal system. As listed in , the gained elastic constants of all Mo1-xLax binary alloys can meet the above criterion, meaning that all calculated Mo1-xLax binary alloys exhibit the mechanical stability under zero pressure and static state condition.
In general, the tetragonal shear modulus C’=(C11-C12)/2 can judge the phase stability of a cubic crystal system when it undergoes a tetragonal deformation [Citation44]. As given in , C’ decreases with the increasing concentration of La in Mo, however, the decreasing trend is relatively small. Therefore, it can be believed that Mo1-xLax (0<x < 0.1) binary alloys should be under dynamically stable with La doping. Such an energetic stability of Mo1-xLax binary alloys could be also explained according to the volume expansion effect. As given above, since the atomic radius of La atom is larger than that of host Mo atom, the alloying element La can expand average volume per atom and increase the average bond strength among atoms in Mo1-xLax binary alloy, which therefore strengthens the ability of Mo1-xLax binary alloy to withstand the shear deformation.
It is well known that both Young’s modulus and shear modulus can be established by the elastic constants in a single crystal system [Citation45]. In different crystal orientations, the tensile stress can be calculated by the product of Young’s modulus and strain, i.e. . For cubic crystal system, the expressions of Young’s modulus have been derived previously in three (<100>, <110> and <111>) orientations [Citation46]. The corresponding formulas are given as
At the same time, the cubic crystal system exhibits two main slip systems, i.e. {110}<111> and {211}<111>, where two shear modulus coincide exactly with each other within the small strain limit, and therefore two shear modulus can be given as
shows both Young’s modulus and shear modulus of Mo1-xLax binary alloy as a function of La content.
Figure 10. The computed Young’s modulus and shear modulus for single crystal Mo1-xLax (0<x < 0.1) binary alloys.
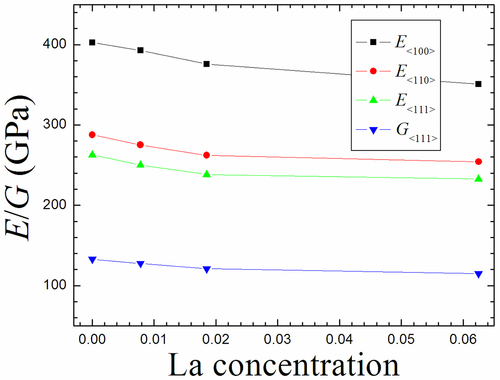
It can be clearly seen that in three orientations, the Young’s modulus displays the same changing tendency and decreases with the increase of La concentration. This indicates that La content is much larger, the required tensile stress is much smaller with the same tensile strain, and therefore the addition of La can diminish the ability of resisting tension/compression within the elastic limit. Similar to the above analysis, Young’s modulus is also applied to judge the stiffness of metal-alloy. Usually, when the Young’s modulus is the larger, there is the greater stiffness of metal-alloy [Citation47]. The Young’s modulus of Mo1-xLax binary alloys as well as perfect Mo obey the following order: Mo>Mo0.9922La0.0078>Mo0.9815La0.0185>Mo0.9375La0.0625, indicating that Mo0.9375La0.0625 alloy possesses the weakest stiffness among these alloys. The shear modulus reveals generally the resistance to shear deformation of metal-alloy, which is relevant with C44. For Mo1-xLax binary alloy, the shear modulus decreases with the increasing La concentration, as shown in . This means that the doping of La exhibits the same influence with the Young’s modulus, i.e. the addition of La reduces the ability of resisting shear deformation within the elastic limit. The shear modulus of Mo1-xLax binary alloys as well as perfect Mo obey the corresponding order: Mo>Mo0.9922La0.0078>Mo0.9815La0.0185>Mo0.9375La0.0625, suggesting that Mo0.9375La0.0625 alloy possesses the weakest resistance to shear deformation among these alloys. Therefore, shear modulus and Young’s modulus tend to be in line.
Note that we have currently carried out first-principles calculations of elastic constants for the ordering binary alloys of Mo1-xLax (0 < x < 0.1). In actual materials, there are random or disordering structures in some local regions, which have been ignored in the present study. Also, the temperature effect has been neglected since the temperature influence has been beyond the scope of first-principles calculations, which can only perform static simulation at absolute zero condition. However, these above mechanical quantities (parameters) could also give a much valuable reference for future researches of Mo1-xLax alloy at a certain temperature scope.
3.5.3. Mechanical parameters for poly-crystal Mo1-xLax binary alloy
For single crystal system, the mechanical quantities cannot well reflect the mechanical properties at a large scale in actual materials, and thus we further study the mechanical parameters of poly-crystal Mo1-xLax binary alloy, which includes Young’s modulus E, bulk modulus B, shear modulus G, Poisson’s ratio v, Anisotropy factor A and B/G. Hill has shown that Voigt and Reuss elastic moduli are the strict upper and lower bound, respectively. Therefore, Hill-averaged bulk modulus B and shear modulus G are gained according to Voigt-Reuss-Hill approximation [Citation48,Citation49]. The corresponding Hill-averaged bulk modulus B of poly-crystal is equal to that of single-crystal and it is given as
Hill-averaged shear modulus G of poly-crystal is written as
and
. Gv (Voigt elastic modulus) and GR (Reuss elastic modulus) denote upper and lower elastic limits of actual poly-crystal constants, respectively. The following averaged E (Young’s modulus), v (Poisson’s ratio), and A (Anisotropy factor) are expressed as
displays the variation trends of Young’s modulus E, bulk modulus B, and shear modulus G for poly-crystal Mo1-xLax alloy as a function of La concentration, and also gives the calculated numerical values in detail.
Figure 11. The computed bulk modulus B, Young’s modulus Eand shear modulus Gof poly-crystal (0 < x < 1.0) binary alloy as a function of La concentration.
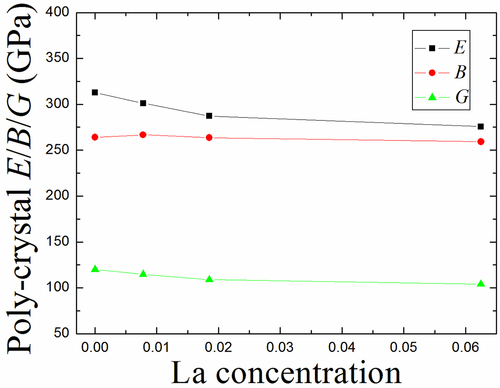
Table 4. The computed bulk modulus B (GPa), Young’s modulus E (GPa), shear modulus G (GPa), Poisson’s ratio (v), Zener anisotropy factor (A) and B/G of poly-crystal Mo1-xLax (0<x < 0.1) alloy.
The bulk modulus B almost keeps invariable with the increasing La content. For example, the bulk modulus alters from 264.1 GPa (perfect Mo) to 259.2 GPa (Mo0.9375La0.0625) and the difference-value between them is only 4.9 GPa. We know that the bulk modulus B is generally applicable for judging the strength of materials [Citation50,Citation51], i.e. it reflects the resistance of material to uniform compression. If the metal-alloy exhibits the smaller lattice parameter, it will possess the larger bond strength, and therefore metal-alloy is hard to be compressed. As given in , the lattice parameter changes from 3.150 Å (perfect Mo) to 3.162 Å (Mo0.9375La0.0625) and the difference-value between them is only 0.012 Å. In other words, Mo1-xLax (0 < x < 0.1) binary alloys keep nearly the same lattice parameters as perfect Mo with the increasing La concentration, leading to that these Mo1-xLax binary alloys nearly have the same bulk modulus and exhibit the similar ductility with perfect Mo.
As mentioned above, the Young’s modulus is often applied to reflect the stiffness of metal-alloy [Citation47]. Similar to the single crystal case, the Young’s modulus of poly-crystal Mo1-xLax alloy also decreases with the increase of La concentration, as shown in . Although the relationship between hardness and Young’s modulus cannot be well identified for different materials, the general trend is that much larger the Young’s modulus, much harder the materials. As seen from , the hardness of poly-crystal Mo1-xLax alloy is gradually reduced with increasing La concentration. For shear modulus G, the changing trend of poly-crystal Mo1-xLax alloy is similar to that of single crystal Mo1-xLax alloy (), and the variation is relatively small.
The ductile/brittle transition is generally applicable for evaluating material properties. Previously, Pugh gave a criterion for the ductile-brittle transition by comparing the B/G value [Citation51]. When the B/G is higher/lower than 1.75, a metal-alloy is in the ductile/ductile region. As given in , the B/G values of all poly-crystal Mo1-xLax alloys are higher than 1.75, demonstrating that these binary alloys are in the ductile phase. At the same time, the B/G of poly-crystal Mo1-xLax alloy is inclined to increase with rising La concentration, suggesting that the ductility of poly-crystal Mo1-xLax alloy increases with increasing La content.
Poisson’s ratio v and Anisotropy-factor A of poly-crystal Mo1-xLax alloy are further calculated as a function of La concentration. Poisson’s ratio is generally applicable for revealing the plasticity of an metal-alloy. Perfect plasticity is good for the machinability of metal-alloy. It can be seen that the Poisson’s ratio increases with increasing La content, consistent with the changing trend of B/G. In other words, Mo0.9375La0.0625 alloy possesses the largest value of 0.323, while Mo0.9922La0.0078 alloy has the smallest value of 0.312. Corresponding, Mo0.9375La0.0625 and Mo0.9922La0.0078 alloys have the best and the worst plasticity, respectively. The Anisotropy-factor A is generally used to reveal the elastic anisotropy of metal-alloys, and it is usually taken as 1 for an isotropic material. The larger the difference-value between A and 1 is, the more serious the elastic anisotropy is. As listed in , the Anisotropy-factors of all these Mo1-xLax alloys as well as perfect Mo are far less than 1, meaning that both perfect Mo and Mo-alloys are wholly anisotropic. In addition, the Anisotropy-factors deviate from 1 farther and farther with increasing La concentration, demonstrating that the elastic anisotropys of these Mo1-xLax alloys is getting more and more serious.
4. Conclusion
Employing first-principles calculations, we have performed the detailed researches for La behavior in Mo including structure, thermodynamic stability, interaction with vacancy and mechanical properties of Mo-La alloy. The alloying element La prefers to stay at SS with the solution energy of 2.86 eV, much lower than the solution energies of 10.57 and 11.19 eV at TIS and OIS, respectively. For three mixed dumbbell structures including < 100>, <110> and <111>, the <111> mixed dumbbell is more stable than both < 100> and <110> mixed dumbbells. The La-La interaction displays attraction and repulsion in short- and long-distance range, respectively. Both 1nn and 2nn shells are most likely to form the La-La clusters since there are relatively larger binding energies, which are 0.43 and 0.33 eV, respectively. The vacancy-La presents a much strong attractive interaction for La at 1nn shell and the binding energy between vacancy and La is 1.33 eV. A weak attractive interaction is present for vacancy-La at 2nn site and the binding energy between vacancy and La is 0.26 eV. The mechanical properties of Mo1-xLax (0 <x < 0.1) binary alloys have been analyzed in detail. (i) For single crystal, the effect of La on C11 is larger than these on both C12 and C44 with increasing La content. The addition of La can strengthen the ability of Mo1-xLax binary alloy to withstand the shear deformation because of the increment of the average bond strength among atoms. (ii) For poly-crystal, the bulk modulus B almost keeps invariable with increasing La content. The changing trend of both Young’s modulus and shear modulus is similar to that of single crystal case. The B/G tends to increase with increasing La concentration, suggesting that the ductility of poly-crystal alloy can be improved. With the increase of La content, Poisson’s ratio presents that the plasticity of poly-crystal alloy is becoming better and better, while Anisotropy-factor displays that the elastic anisotropy of poly-crystal alloy is getting more and more serious.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Byun JM, Hwang SH, Lee S et al. Microstructure control of Mo–Si–B alloy for formation of continuous α-Mo phase. Int J Refract Met Hard Mater. 2015;53:61. doi: 10.1016/j.ijrmhm.2015.03.006
- Yu JL, Li ZK, Zheng X et al. Tensile properties of multiphase Mo–Si–B refractory alloys at elevated temperatures. Eng A. 2012;532:392. doi: 10.1016/j.msea.2011.11.001
- Li WH, Zhang GJ, Wang SX et al. Ductility of Mo–12Si–8.5B alloys doped with lanthanum oxide by the liquid–liquid doping method. J Alloys Compd. 2015;642:34. doi: 10.1016/j.jallcom.2015.04.047
- Dimiduk DM, Perepezko JM. Mo-Si-B alloys: developing a revolutionary turbine-engine material. MRS Bull. 2003;28:639. doi: 10.1557/mrs2003.191
- Marot L, Meyer E, Rubel M et al. JET-EFDA Contributors. J Nucl Mater. 2013;438:S1187. doi: 10.1016/j.jnucmat.2013.01.262
- Miyamoto M, Takaoka H, Ono K et al. Crystal orientation dependence of surface modification in molybdenum mirror irradiated with helium ions. Nucl Mater. 2014;455:297. doi: 10.1016/j.jnucmat.2014.06.030
- Moser L, Steiner R, Leipold F et al. Plasma cleaning of ITER first mirrors in magnetic field. J Nucl Mater. 2015;463:940. doi: 10.1016/j.jnucmat.2014.11.087
- Lee G, Manière C, McKittrick J et al. Consolidation of molybdenum nanopowders by spark plasma sintering: Densification mechanism and first mirror application. J Nucl Mater. 2019;516:354. doi: 10.1016/j.jnucmat.2019.01.028
- Liu G, Zhang GJ, Jiang F et al. Nanostructured high-strength molybdenum alloys with unprecedented tensile ductility. Nat Mater. 2013;12:344. doi: 10.1038/nmat3544
- El-Genk MS, Tournier JM. A review of refractory metal alloys and mechanically alloyed-oxide dispersion strengthened steels for space nuclear power systems. J Nucl Mater. 2005;340:93. doi: 10.1016/j.jnucmat.2004.10.118
- Iorio LE, Bewlay BP, Larsen M. Analysis of AKS- and lanthana-doped molybdenum wire. Int J Refract Met Hard Mater. 2006;24(4):306. doi: 10.1016/j.ijrmhm.2005.10.004
- Yang XQ, Tan H, Lin N et al. The influences of La doping method on the microstructure and mechanical properties of Mo alloys. Int J Refract Met Hard Mater. 2015;51:301–308. doi: 10.1016/j.ijrmhm.2015.04.034
- Cheng PM, Zhang GL, JY Z et al. Coupling effect of intergranular and intragranular particles on ductile fracture of Mo–La2O3 alloys. Mater Sci Eng A. 2015;640:320. doi: 10.1016/j.msea.2015.05.032
- Chen C, Tan W, Wang MP et al. The transverse elongation and fracture mechanism of the upset Mo and Mo–La2O3 bars. Mater Sci Eng A. 2010;527:600. doi: 10.1016/j.msea.2009.09.038
- Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Hafner J Phys Rev B. 1993;47(1):558. doi: 10.1103/PhysRevB.47.558
- Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Furthmüller J Phys Rev B. 1996;54(16):11169. doi: 10.1103/PhysRevB.54.11169
- Alnujaim S, Bouhemadou A, Chegaar M et al. Density functional theory screening of some fundamental physical properties of Cs2InSbCl6 and Cs2InBiCl6 double perovskites. The European Phys J B. 2022;95:114. doi: 10.1140/epjb/s10051-022-00381-2
- Drici L, Belkharroubi F, Boufadi FZ et al. First-principles calculations of structural, elastic, electronic, and optical properties of CaYP (Y = Cu, Ag) Heusler alloys. Emerg Mater. 2022;5:1039. doi: 10.1007/s42247-021-00211-8
- Radja K, Farah BL, Ibrahim A et al. Investigation of structural, magneto-electronic, elastic, mechanical and thermoelectric properties of novel lead-free halide double perovskite Cs2AgFeCl6: first-principles calcuations. J Phys Chem Solids. 2022;167:110795. doi: 10.1016/j.jpcs.2022.110795
- Khireddine A, Bouhemadou A, Maabed S et al. Elastic, electronic, optical and thermoelectric properties of the novel Zintl-phase Ba2ZnP2. Solid State Sci. 2022;128:106893. doi: 10.1016/j.solidstatesciences.2022.106893
- Asma B, Belkharroubi F, Ibrahim A et al. Structural, mechanical, magnetic, electronic, and thermal investigations of Ag2YB (Y = nd, Sm, Gd) full-Heusler alloys. Emerg Mater. 2021;4:1769. doi: 10.1007/s42247-021-00257-8
- Perdew JP, Chevary JA, Vosko SH et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys Rev B. 1996;46:6671. doi: 10.1103/PhysRevB.46.6671
- Perdew JP, Burke K, Ernzerhof M. Perdew, burke, and ernzerhof reply. Phys Rev Lett. 1998;80:891. doi: 10.1103/PhysRevLett.80.891
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865. doi: 10.1103/PhysRevLett.77.3865
- Blochl PE. Projector augmented-wave method. Phys Rev B. 1994;50:17953. doi: 10.1103/PhysRevB.50.17953
- Kresse G, Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B. 1999;59:1758. doi: 10.1103/PhysRevB.59.1758
- Kittel C. Introduction to Solid State Physics. 7th ed. New York: Wiley; 1996.
- Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B. 1976;13:5188. doi: 10.1103/PhysRevB.13.5188
- Methfessel M, Paxton AT. High-precision sampling for Brillouin-zone integration in metals. Phys Rev B. 1989;40:3616. doi: 10.1103/PhysRevB.40.3616
- https://materialsproject.org.
- Domain C, Becquart CS, Foct J. Ab initio study of foreign interstitial atom (C, N) interactions with intrinsic point defects in α -Fe. Foct J Phys Rev B. 2004;69(14):144112. doi: 10.1103/PhysRevB.69.144112
- Kraftmakher Y. Equilibrium vacancies and thermophysical properties of metals. Phys Rep. 1998;299:79. doi: 10.1016/S0370-1573(97)00082-3
- Schafer HE. Investigation of thermal equilibrium vacancies in metals by positron annihilation. Status Solidi A. 1987;102:47. doi: 10.1002/pssa.2211020104
- Muzyk M, Nguyen-Manh D, Kurzydłowski KJ et al. Phase stability, point defects, and elastic properties of W-V and W-Ta alloys. Phys Rev B. 2011;84:104115. doi: 10.1103/PhysRevB.84.104115
- Ohnuma T, Soneda N, Iwasawa MAM. First-principles calculations of vacancy–solute element interactions in body-centered cubic iron. Acta Materialia. 2009;57(20):5947. doi: 10.1016/j.actamat.2009.08.020
- Clouet E, Varvenne C, Jourdan T. Elastic modeling of point-defects and their interaction. Comput Mater Sci. 2018;147:49. doi: 10.1016/j.commatsci.2018.01.053
- Varvenne C, Bruneval F, Marinica M-C, et al. Point defect modeling in materials: coupling ab initio and elasticity approaches. Phys Rev B. 2013;88(13):134102. doi: 10.1103/PhysRevB.88.134102
- Pasianot RC. Self-interstitials structure in the hcp metals: a further perspective from first-principles calculations. J Nucl Mater. 2016;481:147. doi: 10.1016/j.jnucmat.2016.09.021
- Souissi M, Chen Y, Sluiter MH et al. Ab initio characterization of B, C, N, and O in bcc iron: solution and migration energies and elastic strain fields. Comput Mater Sci. 2016;124:249. doi: 10.1016/j.commatsci.2016.07.037
- Olsson P, Klaver TPC, Domain C. Phys Rev B. 2010;81:054102. doi: 10.1103/PhysRevB.81.054102
- Wolverton C. Solute–vacancy binding in aluminum. Acta Mater. 2007;55:5867. doi: 10.1016/j.actamat.2007.06.039
- Wallace C. Phys Status Solidi B. 1970;25:301.
- Zhao JJ, Winey JM, Gupta YM. First-principles calculations of second- and third-order elastic constants for single crystals of arbitrary symmetry. Phys Rev B. 2007;75(9):094105. doi: 10.1103/PhysRevB.75.094105
- Wang J, Yip S, Phillpot SR, et al. Crystal instabilities at finite strain. Phys Rev Lett. 1993;71(25):4182. doi: 10.1103/PhysRevLett.71.4182
- Kelly A, Macmillan NH. Strong solids. 3rd ed. Clarendon, Oxford; 1986.
- Liu YL, Zhou HB, Zhang Y. Ideal mechanical properties of vanadium by a first-principles computational tensile test. J Nucl Mater. 2011;416:345. doi: 10.1016/j.jnucmat.2011.06.027
- Yang JW, Gao T, Gong YSSS. The disproportionation reaction phase transition, mechanical, and lattice dynamical properties of the lanthanum dihydrides under high pressure: a first principles study. Solid State Sci. 2014;32:76. doi: 10.1016/j.solidstatesciences.2014.03.019
- Reuss A, Angew Z. Berechnung der Fließgrenze von Mischkristallen auf Grund der Plastizitätsbedingung für Einkristalle. Z Angew Math Mech. 1929;9:49–58. doi: 10.1002/zamm.19290090104
- Hill R. The elastic behaviour of a crystalline aggregate. Proc Phys Soc A. 1952;65:349. doi: 10.1088/0370-1298/65/5/307
- Meradji H, Drablia S, Ghemid S. First-principles elastic constants and electronic structure of BP, BAs, and BSb. Status Solidi B. 2004;241(13):2881. doi: 10.1002/pssb.200302064
- Pugh SF. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Philos Mag. 1954;45:823. doi: 10.1080/14786440808520496