Abstract
Using refined density functional theory approaches, we investigate the structure and spectroscopic signatures of recently synthesised oxasmaragdyrin dyes. Besides comparisons between theoretical and experimental nuclear magnetic resonance (NMR) chemical shifts, we analyse the electronic excited states of two boron complexes of oxasmaragdyrin in order to rationalise the observed optical spectra. Specifically, we show that the spectral variations induced by complexation with fluorine ions are related to a fine balance between the impact of geometrical deformation and electrostatic effects.
1. Introduction
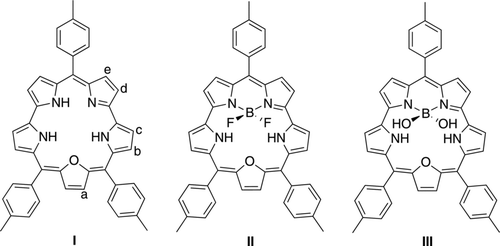
Macrocycles are of key importance in all areas of chemistry. While the most well-known classes of macrocycles, namely porphyrins and phthalocyanins, have been thoroughly investigated, there exist more original π-conjugated macrocyclic structures, e.g. pentaphyrins, hexaphyrins, turcasarin and rosarins [Citation1–Citation3]. Amongst the pentaphyrins, smaragdyrin and its derivatives have been characterised with a large panel of experimental techniques [Citation2, Citation4–Citation12]. These structures were first reported in 1972 [Citation4], as a specific member of the sapphyrin group [Citation13], but their extensive investigation started in 1998 with the works of Sessler [Citation5, Citation6] and later Chandrashekar [Citation7–Citation10] who coupled the smaragdyrin cores to additional optically active moieties (e.g. ferrocene, azobenzene, other macrocycles). Very recently, Rao and Ravikanth have synthesised boron complexes of oxasmaradgyrin (I in Figure ) [Citation11]. They first considered the BF2 adduct (II), that are related to the boron-dipyrromethene (BODIPY) family [Citation14–Citation17]. This therefore offers a possible combination between the valuable optical properties of extended macrocells and the cyanine-like character of BODIPY structures. Such combination is rather common for subporphyrins [Citation18], where the boron center is used to stabilise the structure, but remains, to the best of our knowledge, unique for smaragdyrin. In addition, Rao and Ravikanth have synthesised the corresponding BOH2 compound, that was shown to be a selective and effective fluoride ion sensor. Indeed, III undergoes significant shifts of its main visible bands only when F− anions are added to the solution [Citation11].
Figure 1 Representation of the three investigated smaragdyrin systems. For I, the positions of the relevant protons used in the NMR study are indicated
From the theoretical side, little has been made to investigate the spectroscopic properties of smaragdyrin and its derivatives. Indeed, to the best of our knowledge, the only theoretical investigation of the optical spectra of smaragdyrin was performed more than one decade and relied on a (rather crude by today’s standards) semi-empirical level of theory [Citation6]. Other modelling investigations, such as the one carried out in Ref. [Citation11], are ‘limited’ to structural and energetic data, e.g. information related to the complexation of the F− anion. The present contribution aims to fill this gap by providing insights into the nature of the excited states of the derivatives of I, using robust ab initio approaches. In particular the impact on the complexation of F− on III will be modelled. In particular, we will rely on time-dependent density functional theory (TD-DFT) [Citation19–Citation21], that has become the most widely used tool to compute optical spectra of large molecules, thanks to its extremely valuable effort/accuracy ratio.
2. Computational details
All calculations have been performed with Gaussian 09 [Citation22]. The geometric parameters have been optimised at the PBE0/6-311G(d,p) level [Citation23], and its has been checked that the structures are minima of the potential energy surfaces by computing the vibrational spectra. The nuclear magnetic resonance (NMR) values have been determined thanks to the GIAO-PBE0/cc-pVTZ approach. The chemical shifts were computed using tetramethylsilane as a reference. The solvent effects have been systematically accounted for with the polarizable continuum model (PCM) [Citation24], considering toluene as a solvent for NMR.
The optical absorption spectra have been simulated with TD-DFT using both the PBE0 [Citation23] and the CAM-B3LYP functionals [Citation25]. The latter is a range-separated hybrid that has been shown to be very efficient when significantx charge-transfer transitions take place or when the excited state is highly delocalised [Citation26–Citation29]. For these excited-state simulations, we have used a quite extended basis set, namely 6-311+G(2d,p) that was shown adequate to investigate delocalised excited states [Citation30]. The solvent effects on the optical properties have been determined with the same PCM model [Citation24] using dichloromethane or toluene, consistently with experiment. It is important to stress that these calculations relied on the non-equilibrium linear-response approach of the PCM model [Citation31, Citation32]. There exists more refined environmental schemes [Citation33, Citation34], but their use is mostly suggested for transitions presenting a strong charge-transfer character, which is not the case here (see below).
3. Results and analysis
3.1. NMR spectra
As there are no available X-ray data for the considered compounds, we have chosen to compare computed and measured proton chemical shifts so to confirm the quality of the selected method and our DFT geometries. The results are collected in Table . The theory/experiment discrepancies are of the order of 1 ppm, which is a typical error of DFT [Citation35]. More importantly, the relative trends are perfectly reproduced, i.e. the relative positions of each proton is correctly foreseen in all cases but for the inversion of Hc/Hd in III. Also the most significant variations (increase/decrease) upon complexation with BF2 or B(OH)2 are accurately modelled. This gives confidence in the accuracy of the DFT prediction in the present context. We can therefore safely process to the investigation of the optical signatures.
Table 1 Comparison between theoretical (PBE0, see text) and experimental NMR chemical shifts (ppm) for selected protons (see column ). The experimental data have been taken in Ref. [Citation11]
3.2. Nature of the excited-states
In toluene, II displays a strong Soret-like absorption at 446 nm, a slightly less intense Q-like band centred at 475 nm, as well as other weaker Q-like peaks between 591 and 700 nm [Citation11]. As these latter bands are not essential for the ion sensor properties and have been well analysed for porphyrins, we will not discuss them further. Using PBE0 for II, we obtain Soret-like transition wavelengths at 436 and 425 nm (instead of 426 and 398 nm with CAM-B3LYP, see Table ), in better absolute agreement with experiment, but at the prize of a significant underestimation of the separation between the two peaks. As we are mainly interesting in chemically consistent predictions rather than very accurate reproduction of the position of each band here, we have therefore used solely CAM-B3LYP in the following analysis. The fact that CAM-B3LYP provides a too hypsochromic spectra within the vertical TD-DFT transitions, i.e. overestimates the transition energies, an expected trend for the selected functional (see also computational details) [Citation29].
Indeed, as can be seen in Table , CAM-B3LYP restores the major optical features, with almost perfect separation between the two bands (28 nm in theory versus 29 experimentally) and valid relative intensities (the longest wavelength band being slightly less intense). When the BF2 group is replaced by B(OH)2, the positions of the bands in the 350–500 nm domain of the UV/Vis spectra remains almost unaffected while the absorption intensity slightly decrease [Citation11]. Both effects are correctly reproduced by TD-DFT that foresees negligible variations of the absorption maxima (ca. 3 nm) as well as smaller oscillator strengths in III than in II. In Figure , the density difference plots corresponding to these two bands are displayed for III. Note that this representation is more pertinent than usual frontier orbital plots. Indeed, both the 429 and 395 nm absorption imply more than one pair of molecular orbitals, making interpretation difficult on a molecular orbitals basis. In Figure , it is noticeable that the most intense band is related to large variations of the electronic density all around the pentacyle, whereas the Q-like absorption implies a more localised reorganisation of the electronic cloud, principally centred on the furan ring and vicinal atoms. Experimentally, the addition of F− on III induces significant variations of the optical signatures [Citation11]. More precisely, in CH2Cl2 one notices strong decrease of strong bands at 446 and 475 nm and a concomitant appearance of bands centred at 455 and 487 nm. In other words, the first (second) undergoes a +9 (+12) nm bathochromic displacement. The corresponding TD-DFT values are +9 (+16) nm, again nicely matching experiment. As can be seen in Figure , the nature of the optical transitions are not modified by the interaction with the anion. We have therefore performed simulations using the geometry of the III+F− complex but removing completely the anion. This yielded λmax of 451 and 420 nm. Therefore, the geometrical deformation alone explains (more than) the observed bathochromic shift induced by fluorine ions. When replacing F− by a (negative) point charge, one obtains transitions at 446 and 414 nm, respectively. These values can be compared to the 445 and 404 nm figures reported in Table . Consequently, for the first band, the balance between geometric (batho) and electrostatic (hypso) effects completely explains the measured effect, whereas for the second band, there is a small effect related to the direct interaction between the electrons of the anion and of the pentacycle. This difference is related to the nature of the excited states (see Figure ): for the smallest wavelength absorption F− is vicinal to the regions undergoing a strong electronic reorganisation upon photon absorption.
Figure 2 Reorganisation of the electronic density induced by the photon absorption (in red: increase of density; in blue: decrease of density). Left: free III (top: 429 nm absorption, bottom: 395 nm absorption). Right: F−-complexed III (top: 445 nm absorption, bottom: 404 nm absorption)
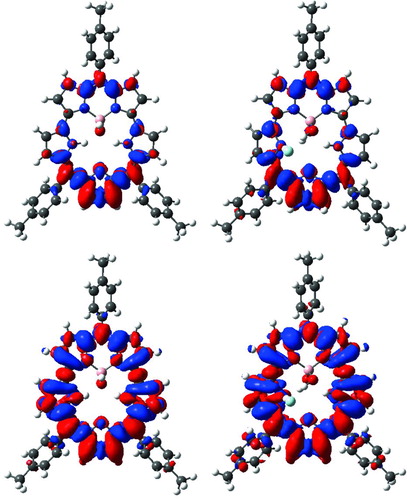
Table 2 Comparison between the computed vertical transition wavelengths (CAM-B3LYP) and experimental λmax for the two most intense bands. Experimental values taken in Ref. [Citation11] and are in toluene () or in CH2Cl2 (). All values in nm, oscillator strengths are given between brackets
4. Conclusions and outlook
In this paper, we have analysed the NMR and UV/Vis signatures of three oxasmaragdyrin derivatives using density functional theory and its time-dependent counterpart, for the ground and excited state properties, respectively. It turned out that the selected level of theory reproduces very satisfactorily the proton shieldings. More importantly, it allowed to analyse the nature of the main absorption bands of the experimental spectra: the two peaks in the 400–480 nm domain imply very different reorganisation of the electronic cloud despite their similar energies and intensities. This hints that smartly localised chemical substitutions of the oxasmaragdyrin will induce significantly different shifts for the two bands, an outcome, that was, to the best of our knowledge, not pointed out previously. On top of that, we have demonstrated that the bathochromic shifts detected upon complexation with fluorine anions can be mainly ascribed to the induced geometrical deformations. Indeed, other effects induce hypsochromic displacements.
We are currently continuing our efforts to characterise pentacycles with the help of theoretical chemistry.
Acknowledgements
Acknowledgements D.J. acknowledges the European Research Council (ERC) and the Région des Pays de la Loire for financial support in the framework of a Starting Grant (Marches - 278845) and a recrutement sur poste stratégique, respectively. This research used resources of (1) the GENCI-CINES/IDRIS (Grant c2012085117); (2) CCIPL (Centre de Calcul Intensif des Pays de Loire) and (3) a local TROY cluster.
References
- Stepien , M. , Sprutta , N. and Latos-Grazynski , L. 2011 . Angew. Chem. Int. Ed. Engl. , 50 ( 19 ) : 4288 doi: 10.1002/anie.201003353
- Pareek , Y. , Ravikanth , M. and Chandrashekar , T. K. 2012 . Acc. Chem. Res. , 45 ( 10 ) : 1801 doi: 10.1021/ar300136s
- Roznyatovskiy , V. V. , Lee , C. H. and Sessler , J. L. 2013 . Chem. Soc. Rev. , 42 ( 5 ) : 1921 doi: 10.1039/c2cs35418g
- Broadhurst , M. J. , Grigg , R. and Johnson , A. W. 1972 . J. Chem. Soc., Perkin Trans. , 1 : 2111 – 2116 . doi: 10.1039/p19720002111
- Sessler , J. L. , Davis , J. M. and Lynch , V. 1998 . J. Org. Chem. , 63 ( 20 ) : 7062 doi: 10.1021/jo981019b
- Gorski , A. , Lament , B. , Davis , J. M. , Sessler , J. and Waluk , J. 2001 . J. Phys. Chem. , A 105 ( 20 ) : 4992 doi: 10.1021/jp004255a
- Venkatraman , S. , Kumar , R. , Sankar , J. , Chandrashekar , T. K. , Sendhil , K. , Vijayan , C. , Kelling , A. and Senge , M. O. 2004 . Chem. Eur. J. , 10 ( 6 ) : 1423 doi: 10.1002/chem.200305558
- Misra , R. , Kumar , R. , Chandrashekar , T. K. and Suresh , C. H. 2006 . Chem. Commun. , 44 : 4584 doi: 10.1039/b610578e
- Misra , R. , Kumar , R. , Chandrashekar , T. K. , Suresh , C. H. , Nag , A. and Goswami , D. 2006 . J. Am. Chem. Soc. , 128 ( 50 ) : 16083 doi: 10.1021/ja0628295
- Gokulnath , S. , Prabhuraja , V. , Sankar , J. and Chandrashekar , T. K. 2007 . Eur. J. Org. Chem. , 2007 ( 1 ) : 191 doi: 10.1002/ejoc.200600719
- Rao , M. Rajeswara and Ravikanth , M. 2011 . J. Org. Chem. , 76 ( 9 ) : 3582 doi: 10.1021/jo200295b
- Pareek , Y. and Ravikanth , M. 2011 . Eur. J. Org. Chem. , 2011 ( 27 ) : 5390 doi: 10.1002/ejoc.201100652
- Bauer , V. J. , Clive , D. L.J. , Dolphin , D. , Paine , J. B. , Harris , F. L. , King , M. M. , Loder , J. , Wang , S. W.C. and Woodward , R. B. 1983 . J. Am. Chem. Soc. , 105 ( 21 ) : 6429 doi: 10.1021/ja00359a012
- Loudet , A. and Burgess , K. 2007 . Chem. Rev. , 107 : 4891 doi: 10.1021/cr078381n
- Quartarolo , A. D. , Russo , N. and Sicilia , E. 2006 . Chem. Eur. J. , 12 : 6797 doi: 10.1002/chem.200501636
- Le Guennic , B. , Maury , O. and Jacquemin , D. 2012 . Phys. Chem. Chem. Phys. , 14 : 157 doi: 10.1039/c1cp22396h
- Ulrich , G. , Ziessel , R. and Harriman , A. 2008 . Angew. Chem. Int. Ed. , 47 : 1184 doi: 10.1002/anie.200702070
- Inokuma , Y. and Osuka , A. 2008 . Dalton Trans. , 0 ( 19 ) : 2517 doi: 10.1039/b719808f
- Runge , E. and Gross , E. K.U. 1984 . Phys. Rev. Lett. , 52 : 997 doi: 10.1103/PhysRevLett.52.997
- Casida , M. E. 1995 . Recent Advances in Density Functional Methods , Edited by: Chong , D. P. Vol. 1 , 155 – 192 . Singapore : World Scientific .
- Jacquemin , D. , Mennucci , B. and Adamo , C. 2011 . Phys. Chem. Chem. Phys. , 13 : 16987 doi: 10.1039/c1cp22144b
- Frisch , M. J. , Trucks , G. W. , Schlegel , H. B. , Scuseria , G. E. , Robb , M. A. , Cheeseman , J. R. , Scalmani , G. , Barone , V. , Mennucci , B. , Petersson , G. A. , Nakatsuji , H. , Caricato , M. , Li , X. , Hratchian , H. P. , Izmaylov , A. F. , Bloino , J. , Zheng , G. , Sonnenberg , J. L. , Hada , M. , Ehara , M. , Toyota , K. , Fukuda , R. , Hasegawa , J. , Ishida , M. , Nakajima , T. , Honda , Y. , Kitao , O. , Nakai , H. , Vreven , T. , Montgomery , J. A. Jr. , Peralta , J. E. , Ogliaro , F. , Bearpark , M. , Heyd , J. J. , Brothers , E. , Kudin , K. N. , Staroverov , V. N. , Kobayashi , R. , Normand , J. , Raghavachari , K. , Rendell , A. , Burant , J. C. , Iyengar , S. S. , Tomasi , J. , Cossi , M. , Rega , N. , Millam , J. M. , Klene , M. , Knox , J. E. , Cross , J. B. , Bakken , V. , Adamo , C. , Jaramillo , J. , Gomperts , R. , Stratmann , R. E. , Yazyev , O. , Austin , A. J. , Cammi , R. , Pomelli , C. , Ochterski , J. W. , Martin , R. L. , Morokuma , K. , Zakrzewski , V. G. , Voth , G. A. , Salvador , P. , Dannenberg , J. J. , Dapprich , S. , Daniels , A. D. , Farkas , O. , Foresman , J. B. , Ortiz , J. V. , Cioslowski , J. and Fox , D. J. 2009 . Gaussian 09 Revision C.01 2009 , Wallingford CT : Gaussian Inc. .
- Adamo , C. and Barone , V. 1999 . J. Chem. Phys. , 110 : 6158 doi: 10.1063/1.478522
- Tomasi , J. , Mennucci , B. and Cammi , R. 2005 . Chem. Rev. , 105 : 2999 doi: 10.1021/cr9904009
- Yanai , T. , Tew , D. P. and Handy , N. C. 2004 . Chem. Phys. Lett. , 393 : 51 doi: 10.1016/j.cplett.2004.06.011
- Peach , M. J.G. , Benfield , P. , Helgaker , T. and Tozer , D. J. 2008 . J. Chem. Phys. , 128 : 044118 doi: 10.1063/1.2831900
- Peach , M. J.G. , Le Sueur , C. R. , Ruud , K. , Guillaume , M. and Tozer , D. J. 2009 . Phys. Chem. Chem. Phys. , 11 : 4465 doi: 10.1039/b822941d
- Goerigk , L. and Grimme , S. 2010 . J. Chem. Phys. , 132 : 184103 doi: 10.1063/1.3418614
- Jacquemin , D. , Planchat , A. , Adamo , C. and Mennucci , B. 2012 . J. Chem. Theory Comput. , 8 : 2359 doi: 10.1021/ct300326f
- Jacquemin , D. and Perpète , E. A. 2006 . Chem. Phys. Lett. , 429 : 147 doi: 10.1016/j.cplett.2006.08.028
- Cammi , R. , Mennucci , B. and Tomasi , J. 1999 . J. Phys. Chem. , A 103 : 9100 doi: 10.1021/jp991564w
- Cossi , M. and Barone , V. 2001 . J. Chem. Phys. , 115 : 4708 doi: 10.1063/1.1394921
- Caricato , M. , Mennucci , B. , Tomasi , J. , Ingrosso , F. , Cammi , R. , Corni , S. and Scalmani , G. 2006 . J. Chem. Phys. , 124 : 124520 doi: 10.1063/1.2183309
- Improta , R. , Barone , V. , Scalmani , G. and Frisch , M. J. 2006 . J. Chem. Phys. , 125 ( 5 ) : 054103 doi: 10.1063/1.2222364
- Bagno , A. 2001 . Chem. Eur. J. , 7 ( 8 ) : 1652 doi: 10.1002/1521-3765(20010417)7:8<1652::AID-CHEM16520>3.0.CO;2-V