
Abstract
Born−Oppenheimer equilibrium structure (r BO e) estimates are reported for benzene and all 12 possible fluorobenzenes, based on geometry optimizations performed at the coupled cluster level of electronic structure theory including single and double excitations augmented by a perturbational estimate of the effects of connected triple excitations [CCSD(T)] and Gaussian basis sets of at least triple zeta quality. Furthermore, accurate semiexperimental equilibrium (r SE e) structures are determined for C6H6, C6H5F, and 1,2- and 1,3-difluorobenzene. They are obtained through a least-squares structural refinement procedure based on equilibrium rotational constants of as many isotopologues as feasible, determined by correcting experimental vibrationally averaged ground-state rotational constants with computed ab initio vibration–rotation interaction constants and electronic g-factors, and using a few structural constraints based on the best r BO e estimates. The r BO e and r SE e equilibrium structures are in excellent agreement with each other for the four semirigid molecules but in almost all cases they differ significantly from previously determined equilibrium structure estimates based on rotational spectroscopy or gas electron diffraction. The nature of deformations of the benzene ring induced by a single fluorine substitution can be characterized as follows: (a) the strongest effect is the pushing of the ipso carbon atom toward the ring center resulting in a deformation at the ipso [by +2.7(1)°] and ortho [−1.7(1)°] CCC angles, (b) a simultaneous decrease in the ortho CC bond length of the benzene ring by 0.009 Å and (c) a decrease of all the CH bond lengths. Additivity relations concerning the F substitution effects are obtained based on the equilibrium structures of all possible fluorobenzenes.
Acknowledgements
The research received support from the Hungarian Scientific Research Fund (OTKA, grant no. NK83583). Contributions to this research by Mr. Béla Mihályi and Ms. Tímea Zoltáni at an early stage of the project are gratefully acknowledged.
Notes
aThe corresponding frozen-core VTZ CCSD(T) results are r(CC) = 1.3975 and r(CH) = 1.0831 Å.
bRef. [Citation52], confirmed during this study using analytic gradients.
cThe all-electron cc-pCVQZ CCSD(T) optimized parameters are r(CC) = 1.3918 and r(CH) = 1.0811 Å.
dSee Equation (Equation5).
eSee Equation (Equation6).
aStandard deviation of the weighted fit, 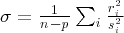 , where ri
is the residual of the ith data, si
is its uncertainty, n is the number of data and p the number of parameters (for a perfect fit, σ = 1).
, where ri
is the residual of the ith data, si
is its uncertainty, n is the number of data and p the number of parameters (for a perfect fit, σ = 1).
ai = ipso, o = ortho, m = meta, and p = para. See also Figure .
b r BO e(I) = CVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
c r BO e(II) = VTZ CCSD(T)_AE + wCVQZ MP2(AE) − VTZ MP2(AE).
aWithout electronic correction.
bWith electronic correction.
cUltrafine grid.
dFine grid.
aReference for the ground state constants.
bFrom the 6-31G* B3LYP cubic force field.
cEquilibrium inertial defect.
dResiduals (obs − calc) of the least-squares fit, Huber weighting.
eFrom the 6-311+G(3df,2pd) B3LYP cubic force field.
aSee text.
bD3 isotopologue excluded, Huber weighting.
aGas electron diffraction (GED) study [Citation65].
bNMR study in a nematic phase [Citation66].
cBest r BO e structure, see Table .
d r SE e structure calculated with the 6-31G* B3LYP force field, see Table .
e r SE e structure calculated with the 6-311+G(3df,2pd) B3LYP force field, see Table .
aGas-phase electron diffraction (GED) studies.
bThe bond lengths are not given because they are not directly comparable to the equilibrium values.
c r BO e(I) = wCVTZ CCSD(T) (AE) + wCVQZ MP2 (AE) − wCVTZ MP2 (AE), see Table .
d r SE e structure calculated with the 6-31G* B3LYP force field, see Table .
aRef. [Citation76].
bCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
cCalculated with the 6-31G* B3LYP force field, Huber weighting, see text.
dCalculated with the 6-31G* B3LYP force field and two predicate observations, see text.
eRef. [Citation75].
aRef. [Citation77].
bCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
cCalculated with the 6-311+G(3df,2pd) B3LYP force field, Huber weighting, see text.
dRef. [Citation78].
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
bRef. [Citation80].
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
bRef. [Citation15].
cEquilibrium structure, Ref. [Citation18].
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
bEquilibrium structure, Ref. [Citation19].
cEffective structure, Ref. [Citation20].
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
bSubstitution structure, Ref. [Citation87].
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
bSubstitution structure of Ref. [Citation91].
cMass-dependent structure of Ref. [Citation91].
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
bEffective structure, Ref. [Citation95].
bSubstitution structure, Ref. [Citation95].
cFrom the spin-spin coupling constants of 19F and 13C nuclei, Ref. [Citation96].
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
aCVTZ CCSD(T)_AE + wCVQZ MP2(AE) − CVTZ MP2(AE).
aCalculated with Equation (Equation8), see text.
bΔ(CCo) = r(CCo) − r(CCbenzene) with r e(CC) = 1.3916 Å and r g(CC) = 1.399 Å [111].
cEmpirical mass-dependent structure from the ground state rotational constants.
aThe numbering of the atoms corresponds to 2,6-difluoro-benzonitrile.
bAssuming that the deformations are additive.
cDeviation from 120°.
dDeviation from the CVTZ CCSD(T)_AE value for benzene, r(CC) = 1.3944 Å, see Table .