
Abstract
A new, general analytical representation of potential energy surfaces (PES) for triatomic molecules is developed. It is based on a novel, angular-dependent form of the Morse potential extended to two bonds. The very compact analytical form needs only a few adjustable parameters while yielding a physically sound description of the global behaviour of the interactions in the entire configuration space. As a first example, the global PES of the lowest adiabatic state of water monomer (H2O) is reviewed. Parameters were adjusted to sets of energy points obtained from ab initio calculations at the coupled cluster and multi-reference configuration interaction level of theory. Experimental vibrational band centres of levels below 5000 cm−1 are correctly reproduced to within 3 cm−1; higher lying levels are less well described than in previously determined PES representations which involved a substantially extended set of adjustable parameters. Spectroscopic and thermodynamic data as well as reaction barriers are overall qualitatively well reproduced. The representation is suitable to describe semi-quantitatively the recombination ![]() as well as the abstraction reaction
as well as the abstraction reaction ![]() in the limit of low collision energies. As a second example, the first global PES of the copper nitrosyl molecule (CuNO) is presented. The equilibrium structure with a bent Cu–N–O connectivity and the dissociation energy match the currently best known values from ab initio calculations (Krishna and Marquardt, J. Chem. Phys.
136, 244303 (2012)) and vibrational band centres agree well with previously published experimental data from matrix isolation spectroscopy for various isotopologues. A high lying metastable state with the linear connectivity N–Cu–O is predicted.
in the limit of low collision energies. As a second example, the first global PES of the copper nitrosyl molecule (CuNO) is presented. The equilibrium structure with a bent Cu–N–O connectivity and the dissociation energy match the currently best known values from ab initio calculations (Krishna and Marquardt, J. Chem. Phys.
136, 244303 (2012)) and vibrational band centres agree well with previously published experimental data from matrix isolation spectroscopy for various isotopologues. A high lying metastable state with the linear connectivity N–Cu–O is predicted.
Acknowledgements
We thank Région Alsace, CNRS as well as the French Ministry for Research for financial support. This work has benefited also from a grant obtained from the French ‘Agence Nationale pour la Recherche’, ANR projet blanc 720 1 ‘DYQUMA’.
Notes
Notes:
a Same value as in column i = 0.
b Same value as in column i = ∞.
c
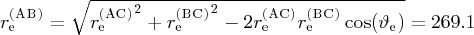 pm.
pm.
d Z (AB) replaced by the z(r (AC), r (BC), ϑ) function, which has the asymptotic value 1.
Notes:
a Harmonic zero-point energy obtained from the sum over all real frequencies.
b
Normal coordinates 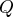 defined with respect to the stationary point; harmonic wavenumbers were calculated from second derivatives
defined with respect to the stationary point; harmonic wavenumbers were calculated from second derivatives 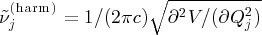 ; negative second derivatives have imaginary wavenumbers; masses as in Table 3.
; negative second derivatives have imaginary wavenumbers; masses as in Table 3.
c Doubly degenerate mode.
Note:
a Level attribution following the suggestion from [24]; we find that, for 63Cu14N16O, the level at 892 cm−1 is a strong resonance between 22 and 2132.
Notes:
a Same value as in column i = 0.
b Same value as in column i = ∞.
c Same value as in column i = iA.
d
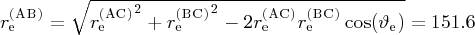 pm.
pm.
e Z (AB) replaced by the z(r (AC), r (BC), ϑ) function, which has the asymptotic value 1.
Notes:
a Level attributions from the analysis of wave functions determined in this work, where available; attributions indicate the most important normal mode product state; when such a clear attribution is not possible, i.e. due to a very strong mixing of modes in the wave function, the ‘polyad’ structure label N j is given, following [8], where N = m + n + k/2; here, m and n are the ‘local mode’ quantum numbers related to the OH stretching overtones, and k is the quantum number of the bending mode (normal mode 2); for the polyad states given in this table, the bending quantum number k is 0 and j = 1, …, N + 1.
b Theoretical values in this column are from [19]; theoretical values from [6] or [54] are not given explicitly here and agree within 0.1 cm−1 or better with the experimental value.
c Values indicated with the superscript c are from [54], where the value 16890 cm−1 is given as the experimental band centre of the 51 state.