
Abstract
The balanced description of ionic and covalent molecular excited electronic states still presents a challenge for current electronic structure methods. In this contribution, we show how the restricted active space self-consistent field (RASSCF) method can be used to address this problem, applied to two dienes in the cis conformation. As with the closely related complete active space self-consistent field (CASSCF) method, the construction of the orbital active space in the RASSCF methodology requires the a priori formulation of a physical or theoretical model of the system being studied. In this article, we discuss how the active space can be constructed in a guided and systematic way, using pairs of natural bond orbitals as correlating partner orbitals (oscillator orbitals) and semi-internal correlation. The resulting balanced description of the covalent and ionic valence excited states – with the ionic state correctly lower in energy at the Franck–Condon geometry – is suitable to study the photochemistry of these and other molecules.
1. Introduction
Photochemical reaction paths [Citation1–4] represent a challenge to theoretical chemistry methods because they generally involve a changing balance between open vs. closed shell configurations and covalent vs. ionic valence bond (VB) structures. Computational chemistry procedures need to be able to represent these very different bonding situations with the same relative accuracy. Most complete active space self-consistent field (CASSCF) treatments that use relatively small active spaces poorly represent ionic VB structures in preference to covalent structures [Citation5–7]. Density functional theory (DFT) methods can have problems representing charge transfer [Citation8] and conical intersections (CIs) [Citation9]. CASPT2 can to a good extent improve the CASSCF result, but because the orbitals are not optimised, orbital relaxation effects are included only at the perturbative level which can be a problem if the CASSCF reference wavefunction gives a poor description of the ionic electronic state. In this work, we will explore an alternative route for improving the CASSCF result by judiciously extending the configuration interaction expansion using the restricted active space self-consistent field (RASSCF) method [Citation10–14], which includes full orbital optimisation and for which analytical gradient techniques are available. This approach involves only a small increase in computational complexity with the benefit of giving new insights into the factors influencing the electronic correlation effects of the system. The aim of this contribution is not to focus on accurate spectroscopic calculations achieved by highly correlated methods. Instead, we aim at a qualitatively correct and balanced description which allows for the study of potential energy surfaces relevant for photochemistry and subsequent on-the-fly dynamics calculations, for which analytic gradients techniques are indispensable.
We will be focussing our attention on the photochemistry of two well-studied conjugated cis-diene systems: cis-butadiene (BD) [Citation7,Citation15–22] and cyclopentadiene (CPD) [Citation6,Citation23–26]. In this context, the photochemical reaction path can be understood as the intrinsic reaction path [Citation27] starting in the Franck–Condon (FC) region of the spectroscopically bright excited state (1B2 in our case). For both molecules, the FC region is zwitterionic, yet the reaction funnel (the last CI on the reaction path) involves two electronic states of covalent character. The reaction path therefore consists of covalent–ionic photoexcitation, an initial reaction path on an ionic state, a transition from an ionic to covalent electronic character (associated with a nearby ionic–covalent CI) and finally a non-planar covalent–covalent CI [Citation1,Citation23] giving access to the ground state surface where different photoproducts can form [Citation15,Citation16,Citation23]. In this work, we will be concerned solely with the portion of the reaction path taking place on the S1 surface. The correct description of such an intricate reaction path needs a balanced treatment of both ionic and covalent electronic states, which in the case of CIs needs to be obtained in a single calculation.
can be understood as the intrinsic reaction path [Citation27] starting in the Franck–Condon (FC) region of the spectroscopically bright excited state (1B2 in our case). For both molecules, the FC region is zwitterionic, yet the reaction funnel (the last CI on the reaction path) involves two electronic states of covalent character. The reaction path therefore consists of covalent–ionic photoexcitation, an initial reaction path on an ionic state, a transition from an ionic to covalent electronic character (associated with a nearby ionic–covalent CI) and finally a non-planar covalent–covalent CI [Citation1,Citation23] giving access to the ground state surface where different photoproducts can form [Citation15,Citation16,Citation23]. In this work, we will be concerned solely with the portion of the reaction path taking place on the S1 surface. The correct description of such an intricate reaction path needs a balanced treatment of both ionic and covalent electronic states, which in the case of CIs needs to be obtained in a single calculation.
A fundamental aspect of the photochemical reaction path is the energetic ordering at the FC geometry of the three electronic states involved in the process, with the zwitterionic bright state (whose wavefunction transforms according to the B2 irreducible representation of the C2v point group) lower in energy than the covalent dark state (with the A1 symmetry character) [Citation6,Citation7,Citation22,Citation28]. The CASSCF method when applied to two π bonds, involving a four electrons and four π orbitals active space, fails to reproduce the correct order of the states at the FC geometry [Citation19]. The overestimation of the B2 state vertical excitation energy (above the 2A1 state) implies that a more careful description of electron correlation is necessary. It is the purpose of this article to show that one can describe BD and CPD photochemistry, from the FC region through two CIs to the ground state, using RASSCF, with an active space selected according to a physical model that can realistically describe the differences in electron correlation between both excited states as a function of molecular geometry.
The RASSCF method can be seen as an extension of CASSCF where the active space is divided into three sub-spaces: RAS1, RAS2 and RAS3. As in CASSCF, all possible configurations of electrons within the RAS2 orbitals are included in the multi-configurational self-consistent field (MCSCF) expansion, which is extended by configurations which feature a limited number of vacancies in the RAS1 orbitals (in this study, we will allow for single vacancies only), and a limited number of occupancies in the RAS3 orbitals (one or two in our case).
The RASSCF method suffers from similar conceptual difficulties to CASSCF because one needs to choose an active space before the calculation begins. This needs to be done using some physical or theoretical model. Two fundamental ideas will guide us in improving the treatment of electron correlation beyond CASSCF(4,4) for these systems: the first is the notion of correlating orbital pairs, or oscillator orbitals in Boys’ terminology [Citation29,Citation30], which will assist in the choice of orbitals to include in the active space, and the second is the notion of semi-internal correlation, first discussed by Sinanoğlu [Citation31,Citation32] for atoms, which will determine the order to which the configuration interaction expansion should be extended. (For other ways of choosing a RASSCF active space, see [Citation33] and [Citation34].)
Correlating orbital pairs/oscillator orbitals have the property that they are localised in the same region of space as the occupied orbitals but have higher numbers of nodes, and their inclusion in a configuration interaction expansion provides a systematic way to account for electron correlation [Citation35]. The bonding and anti-bonding pairs of natural bond orbitals (NBOs) of Weinhold [Citation36] have this characteristic. NBOs are built by combining natural atomic orbitals (NAOs) [Citation37,Citation38]. The NAOs are obtained from the (block) diagonalisation of the interatomic part of the one electron density matrix. Note that ‘strongly’ occupied NBOs are often used in the analysis of chemical bonding [Citation39]. In this work, in addition to the strongly occupied NBOs, we use the more weakly occupied NBOs to construct the active space of a RASSCF wavefunction forming oscillator orbital pairs. Here, this allows us to extend the active space to describe the important correlation effects that differ between ionic and covalent states.
In a multi-reference configuration interaction expansion, if one allows only single excitations from the ‘closed shell sea’ (RAS1 in RASSCF), then one recovers the so-called semi-internal correlation of Sinanoğlu. Strictly speaking, configurations including single excitations from RAS1 coupled with single excitations from RAS2 recover semi-internal and spin polarisation effects. If one includes double excitations from the closed shell sea, then it is dynamic correlation energy that is being recovered. The effect of semi-internal correlation on the relative energies of excited states with different bonding characteristics (eg. covalent vs. ionic) is strongly structure dependent, and thus critically affects the shape of potential energy surfaces (i.e. the correlation of a σ electron with a π electron will be very different for a covalent as opposed to a zwitterionic state [Citation40]). In contrast, dynamic correlation is much less structure dependent because it is approximately the same for each state.Footnote1
In Section 2, we will illustrate how the concepts of correlating orbital pairs and semi-internal correlation can be used to build a RASSCF active space for these and related systems using NBOs. Section 3 offers some technical specifications for the calculations. In Section 4.1, we show how the energy of the B2 state of BD and CPD at the FC geometry progressively lowers as the active space is increased to include different correlation effects, and in Section 4.2, we show how the RASSCF methodology can provide information about the photochemical reaction path away from the FC region. The article ends with concluding remarks in Section 5.
2. Constructing an active space from NBOs
In this section, we illustrate the practical construction of the RASSCF active space using the correlating orbital pairs idea discussed previously. As mentioned in the introduction, we use NBOs as the practical implementation of the correlating pairs/oscillator orbital concept for the choice of orbitals in the active space.
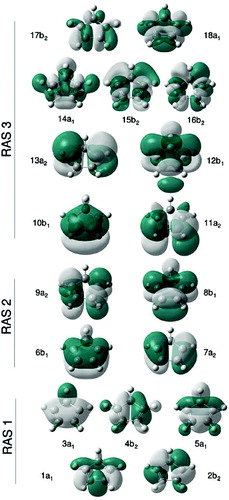
In order to describe π → π* transitions involved in the lower valence electronic states, the most important orbitals to include in the (RAS2) active space are the four 2p π orbitals involving the unsaturated carbon atoms of BD and CPD which are used in a traditional CASSCF(4,4) calculation. In the present case, we choose these orbitals from two bonding and anti-bonding NBO pairs corresponding to the two formal double bonds in the ground state of the molecule. As can be seen from and , orbitals 1π and 4π, along with 2π and 3π, fulfil the criteria of pairs of correlating orbital partners, in the sense that they occupy the same region of space but possess a different nodal structure. This is a different but equivalent way of presenting the CASSCF(4,4) minimal π active space.
Figure 1. NBOs used to build the largest RASSCF active space used in this study for BD at a C2v geometry, using the extended basis set.
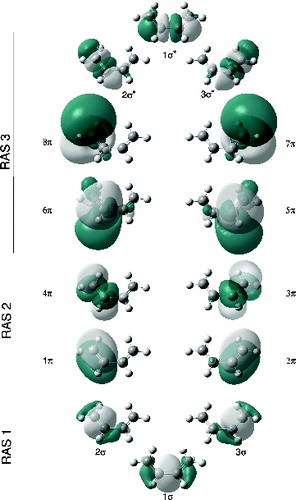
Figure 2. NBOs used to build the largest RASSCF active space used in this study for CPD at a C2v geometry, using the extended basis set.
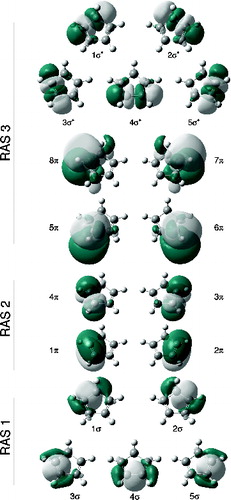
A CASSCF(4,4) active space cannot represent both the zwitterionic state (B2) and the covalent state (A1) adequately for two reasons.(1) It misses the differential (ionic vs. covalent) effect of σπ correlation [Citation5,Citation22,Citation40–42] and (2) the orbital description of the ionic state is inherently much more diffuse than the covalent state [Citation5,Citation19,Citation22,Citation42,Citation43] (because of configurations involving charge redistribution); therefore, it requires both 2p-type atomic orbitals and a set of four oscillator orbitals built from 3p carbon orbitals with additional nodes above and below the molecular plane, to describe the correlation energy of the π system.
The first effect, the differential (ionic vs. covalent) effect of σπ correlation on the ionic state, can be understood as arising from the mutual polarisation of the π and σ electrons. This is a semi-internal correlation due to the σ frame orbitals and, as alluded to in the introduction, the structure dependence of this effect can affect the shape of the potential energy surfaces. The semi-internal correlation effect will be included by adding to the RAS1 sub-space the σ NBOs localised on the inter-atomic axes between carbon atoms (three for BD and five for CPD), and the corresponding σ* in RAS3, and allowing only for single vacancies in RAS1 together with single excitations into RAS3. Once more, {σ, σ*} orbital pairs constitute localised oscillator orbitals.
The second effect, correlation of the π system itself [Citation5–7,Citation19,Citation22], can be recovered by adding to the active space a set of orbitals which will act as oscillator orbitals for the π system, which in this case will correspond to 3p-type π orbitals providing the extra nodal structure (analogous to the 3d double-shell effect in first row transition metal species [Citation44]). It is important for the basis set to have enough flexibility to describe such orbitals, which we achieved by extending a conventional Pople-type basis set and specifically adding a set of more diffuse (and penetrating) 3p orbitals (see Section 3 for details) which already have the desired extra node. π oscillator orbitals are used by adding to the RAS3 sub-space four largely non-bonding 3p-type orbitals centred on the carbon atoms from the NBO set (see and ). The fact that such NBOs give rise to the desired oscillator orbital pairs can perhaps be better appreciated by inspecting the orbitals obtained after the MCSCF procedure shown in and . Since we are aiming to recover π dynamical correlation, double excitations from 2p-type orbitals in RAS2 into 3p-type orbitals in RAS3 must be included; thus, whenever 3p-type orbitals are used, we shall allow double excitations into RAS3 (keeping single vacancies in RAS1).
Figure 3. Orbitals forming the largest RASSCF active space used in this study for BD, built from the initial NBOs in .
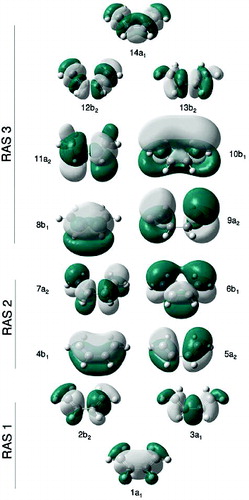
Figure 4. Orbitals forming the largest RASSCF active space used in this study for CPD, built from the initial NBOs in .
An alternative way of improving the dynamical correlation of the π system is to include the extra 3p-type π orbitals into the RAS2 space, as it was done in Ref. [Citation19] (in this latter case selected from the Hartree–Fock molecular orbitals, instead of NBOs as in the present description). However, this results in a calculation with a higher number of terms in the MCSCF expansion with no significant improvement of the resulting excited state energies, since, as it will be shown subsequently, major contributions to the description of the lower valence excited states arise from σπ semi-internal correlation.
The two elements of the physical model for the active space used here are therefore (1) σπ semi-internal electron correlation with {σ, σ*} orbital pairs limited to single excitations and (2) dynamic correlation of the π electrons using a set of π 3p oscillator orbitals in RAS3.
We make a final practical note on the use of NBOs to build the RASSCF active space. As these orbitals are localised and do not have the same symmetry as the molecule, it is desirable to canonicalise them in some fashion before stating the RASSCF procedure. This could be done by block diagonalising some one-electron operator.Footnote2
3. Computational details
All the calculations were carried out with a development version of Gaussian [Citation45], with its RASSCF implementation described in Ref. [Citation12]. Unless stated otherwise, all calculations were performed state-averaging between all the first three valence states. Unlike in Ref. [Citation19], no symmetry restrictions were imposed on the wavefunction.
Throughout the study, two different basis sets were used: a standard Pople 6-31G* basis set and an extended basis set [Citation19] obtained from the former by explicitly adding 3p atomic orbitals to carbon atoms (all BD carbon atoms, but only to the four unsaturated carbon atoms of CPD). The 3p functions used were taken from the 6-31G basis set of silicon, which in this case consists of a contracted p function with a radial node and an uncontracted p function without a radial node. The use of this extended basis set will be indicated by the ‘+3p’ notation.
The ground-state equilibrium geometry was optimised at the CASSCF level with an active space of four electrons in four orbitals with no state-averaging, using the standard basis set, yielding a C2v equilibrium geometry for both BD and CPD. (Note that bigger CASSCF active spaces [Citation46] and highly correlated methods [Citation47] have suggested that the planar C2v BD geometry is unstable with respect to deformation of the central bond backbone dihedral and that the local minimum corresponds to a distorted structure. As the differences in electronic character between electronic states are more marked in the symmetric geometry and can be given a symmetry label, we will consider a C2v initial BD structure as the FC geometry.)
The initial set of NBOs was obtained by diagonalising the one-electron density matrix of the Kohn–Sham orbitals of a DFT calculation with the B3LYP functional.
Evaluation of the gradients was performed without solving the coupled-perturbed equations in the MCSCF cycle [Citation48],Footnote3which introduces a small correction to the gradient neglected in this study. These corrections increase with the energy gap between states, and therefore are expected to be more important at the FC region and negligible at CIs. They are also more important if the change in orbital relaxation with change in geometry is very different for the states being averaged over. In order to test the effect of the coupled-perturbed equations on the calculation of the gradient, CASSCF(4,8) calculations including all π orbitals in were performed for BD with and without these corrections. The magnitude of the difference of the two gradient calculations on the B2 state at the FC geometry is about 3% of the gradient magnitude.
The intrinsic reaction paths presented in Section 4.2 were calculated using the damped velocity Verlet algorithm [Citation49], which only requires gradient information, with the velocity being set to 0.01 Bohr fs− 1 at each step.
4. Results and discussion
4.1. Excitation energies
4.1.1. cis-butadiene
In this section, we study how the relative energy of the three lower valence singlet states of BD varies as the active space is gradually enlarged to include different correlation effects described in Section 2 via the RASSCF procedure. In this work, the notation RASSCF(a,b,c) will be used to define the active space size, with a, b and c representing the number of orbitals included in the RAS1, RAS2 and RAS3 sub-spaces, respectively.
shows the effect of adding each of the ingredients in a stepwise fashion. At the CASSCF(4,4) level, no σπ correlation is included, and the result shows a highly destabilised ionic 1B2 state. The effect of the σ polarisation is included in the RASSCF(3,4,3) calculation, where single excitations are allowed from RAS1 σ into RAS3 σ* orbitals. Even if the energetic order of the states in this case is still not correctly reproduced, the inclusion of the σ orbitals into the RAS1 and RAS3 sub-spaces was effective in bringing the 1B2 state down in energy.
Figure 5. cis-butadiene singlet electronic state energies at the ground-state equilibrium geometry. The experimental value corresponds to the maximum of a broad UV absorption band [Citation50].
![Figure 5. cis-butadiene singlet electronic state energies at the ground-state equilibrium geometry. The experimental value corresponds to the maximum of a broad UV absorption band [Citation50].](/cms/asset/d7bc97c2-5c76-4e77-9bac-09739535c502/tmph_a_1025880_f0005_oc.jpg)
Using the extended basis set in the RASSCF(3,4,3) calculation, a better description of the polarisation of the π system by the σ frame is achieved. The improved description of the orbital polarisation effect further stabilises the 1B2 state, which gets closer in energy to the 2A1 but nevertheless without inverting the excited-state ordering.
Increasing the size of the RAS3 active space by introducing the four additional π orbitals from the 3p shell and including double excitations into RAS3 orbitals (while keeping only single excitations from RAS1), thus improving the description of dynamical correlation of the π system, has proved to be fundamental to reproduce the correct energetic state ordering in BD. While the introduction of double excitations to RAS3 has a little effect on the σ* orbitals which conserve similar values of occupation number, it has an important effect on the increase of π 3p occupation number (note that the 2A1 state has an important contribution from a doubly excited π → π* configuration), stabilising the active space. In this RAS(3,4,7)+3p calculation, the 1B2 electronic state is significantly stabilised and lies 0.38 eV (8.6 kcal mol− 1) below the 2A1 state.
The vertical energy values in illustrate how the 1B2 state is gradually stabilised by the stepwise inclusion of the effects discussed in Section 2, and approaches the position of the maximum UV absorption value [Citation50]. Even if our best estimate for the excitation energy to the 1B2 state is about 0.8 eV (18 kcal mol− 1) above the maximum absorption band and the best estimates of highly correlated methods [Citation7,Citation22,Citation51,Citation52], in this work we are aiming at a balanced, and qualitatively correct, description of the three valence states in a single calculation using a modest size basis set suitable for geometry optimisation, as well as an illustration of the factors influencing the electron correlation in these systems, instead of a high-accuracy calculation. Regarding the low oscillator strength transition to the valence 2A1 state, to our knowledge no experimental information is available about the energy of this transition at the ground-state equilibrium geometry, but equation of motion coupled cluster [Citation52] and CASPT2 (with an enlarged π active space CASSCF reference wavefunction) [Citation7] calculations estimate it to be 6.8 eV and 6.04 eV, respectively. In our RASSCF(3,4,7) calculation, we obtained a vertical excitation energy of 6.76 eV, in good agreement with the former value. As can be seen in , the relative energy of the 2A1 state increases by about 0.3 eV by adding the semi-internal correlation due to σ electrons, while improving π electron correlation has the opposing weaker effect of lowering the state's energy.
Although the RASSCF(3,4,7)+3p calculation is fully converged and gives a qualitatively good result, one of the π orbitals in the RAS3 active space, the 9a2 orbital shown in , has a low occupation number (about 0.003), revealing its contribution to the wavefunction is small. In practice, the presence of this orbital makes the active space unstable upon geometry deformation (the orbital is often rotated out of the active space in the MCSCF procedure). We thus removed this orbital from the active space in a RASSCF(3,4,6)+3p calculation, obtaining only marginally different results from RASSCF(3,4,7)+3p. Note that the former calculation cannot be done a priori from NBOs such as described in Section 2, as the removed orbital is a linear combination of all the π orbitals involving the four carbon atoms in the molecule. The RASSCF(3,4,6)+3p active space is the one used subsequently in Section 4.2 to explore the S1 state potential energy surface.
4.1.2. Cyclopentadiene
As with BD in Section 4.1.1, here we study the energy of the three lowest singlet valence states of CPD (which differs from BD by only the CH2 group closing the ring), as a function of different active space configurations in a RASSCF procedure. The results are summarised in .
Figure 6. Cyclopentadiene electronic energy values at the ground-state equilibrium geometry. The experimental value corresponds to the maximum absorption position on the UV spectrum [Citation24].
![Figure 6. Cyclopentadiene electronic energy values at the ground-state equilibrium geometry. The experimental value corresponds to the maximum absorption position on the UV spectrum [Citation24].](/cms/asset/9a82ec88-b964-45a3-81f4-da79c86ad84c/tmph_a_1025880_f0006_oc.jpg)
The first calculation performed on CPD to introduce the σ semi-internal correlation effect, including the σ bonding and anti-bonding orbitals in the RAS1 and RAS3 sub-spaces, is a RASSCF(5,4,5) calculation (i.e. the active space increases from RASSCF(3,4,3) in BD with the inclusion of the extra σ and σ* orbitals in the ring, as can be seen in ). Even if BD and CPD molecules present very similar structures, the inclusion of this first effect for CPD already highlights a difference between the two in the treatment of the electronic structure, with the 1B2 state lying 0.3 eV (7 kcal mol− 1) below 2A1. This shows how the polarisation of the σ frame already corrects the result previously obtained at the CASSCF(4,4) level, which was showing the 1B2 state more than 1 eV (20 kcal mol− 1) above the covalent state.
Further extending the size of the basis set to increase π orbital flexibility, keeping the same RASSCF(5,4,5) active space, results in a further stabilised 1B2 state, now standing 0.45 eV (10 kcal mol− 1) below 2A1, as shown in . This latter result was further improved by explicitly introducing four 3p-type orbitals into the RAS3 active space (see ), and by allowing double excitation into this sub-space. The π dynamical correlation effect further stabilises the 1B2 state, bringing it closer to the experimental excitation energy [Citation24]. In this case, the stabilisation applies equally to the 1B2 and 2A1 states, as their energy difference does not significantly change from the previous calculation. These results suggest that, in contrast with BD, in CPD the π dynamical correlation is less important than the semi-internal correlation due to σ orbitals in the description of the relative stability of the ionic and covalent excited states, even if it contributes to lowering the vertical excitation energy, bringing it closer to the experimental value.
Similar to the BD case, our results for CPD show computed vertical excitation energies to the 1B2 state more than 0.65 eV (15 kcal mol− 1) higher than the ones predicted by coupled-cluster [Citation26,Citation53] and CASPT2 (with a minimal four electrons in four orbitals [Citation25,Citation53,Citation54], or a more extended active space [Citation6], CASSCF reference wavefunction) methods, which are in better agreement with the maximum absorption position of the UV spectrum [Citation24] (see Ref. [Citation26] for a brief discussion on the difference between maximum absorption intensity and vertical excitation energy in this system). The 1A1 →2A1 transition has a very low oscillator strength and is thought to spectrally overlap with the stronger 1A1 →1B2 transition [Citation55]. Although no definite experimental assignment has been done, and one resonance Raman analysis [Citation55] of the spectrum hints at a 2A1 energy value below 1B2, most high-level theoretical estimates [Citation6,Citation25,Citation53,Citation54] point to a 2A1 state above 1B2 by a variable amount, usually with an excitation energy from 1A1 above 6.3 eV (145 kcal mol− 1). In our best calculations, the excitation energy to 2A1 is 6.81 eV (157 kcal mol− 1), which is within the range obtained in other computations (note, however, that because our excitation energy to 1B2 is overestimated, our 1B2–2A1 energy gap is somewhat underestimated). As in the case of BD, σ semi-internal correlation destabilises the 2A1 state relative to the ground state, while π correlation weakly stabilises it.
The results obtained for CPD highlight that some differences arise when comparing the electronic structure to BD. Since the only structural difference between the two molecules is the CH2 group that closes the cyclopentadiene unit into a ring, to further test the effect of this group on the electronic structure of the molecule, we performed a set of tests on CPD in which the backbone σ orbitals involving the CH2 group, namely orbitals 1σ, 2σ and corresponding anti-bonding orbitals in , were not included in the active space. A first calculation performed at the RASSCF(3,4,3) level (not shown in the figure) shows a result similar to the one obtained in the BD context, as only including the σ orbitals relative to the butadiene unit in CPD does not result in the correct excited state energy ordering, and the 1B2 state is 0.20 eV (4.6 kcal mol− 1) above the 2A1 state. The addition of the extended basis set makes the ionic and covalent states almost degenerate and only increasing the RAS3 active space by adding the four 3p orbitals brings the 1B2 state below the covalent one. At the RASSCF(3,4,7)+3p level, the energetic order of the states is correctly reproduced (see ), even though the energy gap between the ionic and covalent states is small and, by comparison, is even more narrow than the one obtained at the same level of theory on BD. Semi-internal correlation due to the CH2 group σ orbitals is thus important in the description of the excited electronic structure description for CPD.
As in the case of BD, the largest active space used for CPD proved unstable to structural deformation; therefore, in the studies of the S1 potential energy surface in Section 4.2, the RASSCF(5,4,5)+3p active space has been used as it is sufficient to describe the correct state ordering in the FC region.
4.2. Potential energy surface features
In Section 4.1 we have shown that our RASSCF approach achieves a balanced description of the ionic and covalent excited states of BD and CPD in the FC region. With an active space that can describe σ semi-internal correlation and π dynamical correlation, the ionic excited state is correctly below the covalent excited state at the FC geometry. In this section, we extend our treatment to nuclear geometries away from the FC region, making use of the analytical gradient techniques available for MCSCF wavefunctions, such as RASSCF wavefunctions, to explore the features of the photochemistry of these molecules, describing their reaction path on the S1 potential energy surface and its CIs. In this section, BD and CPD are treated with a RASSCF(3,4,6)+3p and a RASSCF(5,4,5)+3p active space, respectively.
The photochemistry of BD and CPD will be initiated on S1, as it presents an ionic character in the FC region and is the one populated by photoexcitation. shows an energy profile along the photoreaction path on the S1 state both for BD and for CPD starting at the FC geometry. As the gradient transforms according to the totally symmetric representation of the molecular point group, the motion along the mass-weighted steepest descent path conserves symmetry and both molecules stay in a C2v configuration (solid lines in ). As seen in , the deformation along the path changes the bond length alternation (BLA), with extension and contraction of formally double and single bonds, respectively (see ), which in CPD involves mostly the butadiene sub-unit in agreement with results in Ref. [Citation25]. Along this path, relatively close to the FC region, both systems reach a CI (or very narrow avoided crossing) between S1 and S2, located 0.33 eV (7.6 kcal mol− 1) and 0.57 eV (13 kcal mol− 1) below the FC region for BD and CPD, respectively (see Figure ), where the electronic character of S1 changes from B2 to A1.
Figure 7. Electronic energy profile along the reaction path on S1, starting from the FC geometry, for BD (left) and CPD (right). Thick lines follow the gradient and conserve the C2v symmetry. These lines are styled as in and according to the nature of the electronic state 1A1 (fine dash), 1B2 (large dash) and 2A1 (solid). Thinner lines correspond to reaction paths that deviate from the C2v symmetry, where the electronic states start to mix. The thin line colour reflects the predominant state character. The reaction coordinate is expressed in mass-scaled displacement units, Ångstrom times squared root atomic mass units.
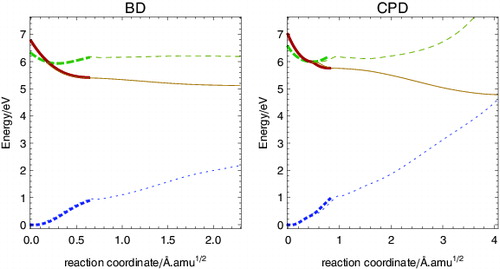
Figure 8. Geometry deformations along the S1 reaction path for BD (left) and CPD (right). As in , thick lines correspond to a path in C2v while thin lines deviate from it, and are styled according to the S1 electronic character. BLA deformation of the butadiene unit is measured from distortions with respect to the ground-state equilibrium geometry: extension of double bond 1 − contraction of single bond + extension of double bond 2. Disrotatory distortion is measured by the dihedral angle of the hydrogen atom at the end of the butadiene unit and the three nearest carbon atoms of this unit. While for BD Cs symmetry is conserved along the path (see the backbone dihedral on the lower panel) and both ends of the molecule describe similar motions, for CPD, the symmetry is lost and the motion of both ends of the molecule differs: different lines correspond to the dihedral of atom numbers 5 and 9 in Figure .
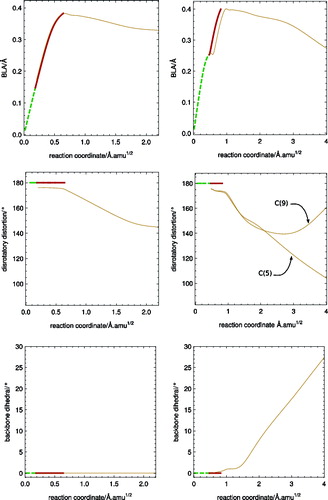
Figure 9. Graphical representation of the forces (negative the gradient) acting on both molecules in the S1 FC region (a and b). The direction of the derivative non-adiabatic coupling in the vicinity of the S2/S1 CI along the reaction path (c and d).
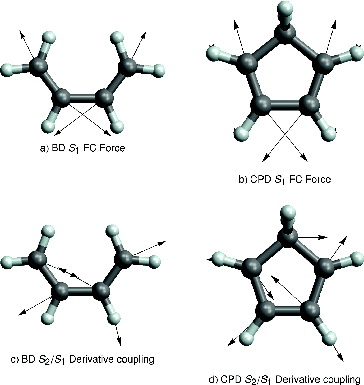
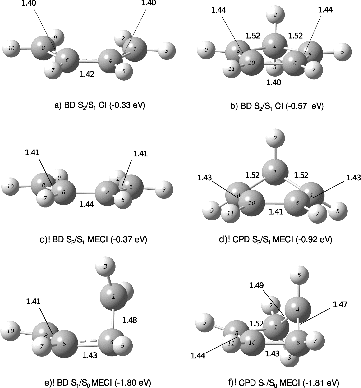
Figure 10. CPD and BD MECIs (c, d, e, f) and C2v CIs located along the steepest descent path on S1 (a, b). The labels on the bonds refer to the bond lengths. The numbers in brackets are the energy differences with respect to the S1 state at the FC geometry. (The cartesian coordinates of these structures are available as Supplemental data.)
Further deforming the molecules on S1 along the (totally symmetric) gradient, a stationary point on a generally rather flat region of the S1 surface is reached, which has some local instability with respect to the non-totally symmetric disrotatory motion for both BD and CPD, and is also unstable with respect to non-totally symmetric pyramidalisation of the CH2 group of CPD. However, one should be cautious about overstating the importance of this path based only on gradient information, as both molecules during dynamics can be expected to distort and deviate from the mass-weighted steepest descent path before reaching this point of the potential energy surface.
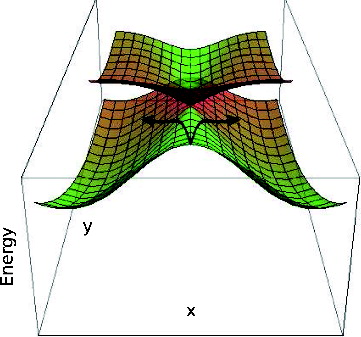
The first reason for deviations from the mass-weighted steepest descent path on S1 is the existence of an S2/S1 CI along this path. schematically shows the potential energy surfaces along the branching space coordinates, for either of the molecules on the S1 ionic state approaching the CI. In both cases, the C2v mass-weighted steepest descent path close to the CI forms a ridge on the S1 potential energy surface, with the motion along non-adiabatic derivative coupling lowering the symmetry of the structure and the energy of S1. This is general for systems approaching a CI from the lowest electronic state [Citation56], and is not specific to the molecules studied here. In the vicinity of a CI, there will be a direction in the coordinate space, orthogonal to the direction of approach, which lifts the degeneracy by lowering the energy of the lower lying electronic state (see Figure ). As the non-adiabatic derivative coupling is always tangential to the direction of approach of a CI [Citation57], the molecules will deform along the direction of this vector. The magnitude of this deformation will depend on how deep are the potential energy ‘valleys’ on the side of the CI (see Figure ). In the case of both molecules studied, the deformation along the non-adiabatic derivative coupling is an in-plane deformation changing bond angles between neighbouring carbon atoms represented in (c) and (d), which results in a small stabilisation of S1. Therefore, although some degree of in-plane asymmetric deformation can be expected, it should be relatively small, and the initial stages of the dynamics should be dominated by the BLA character of the gradient [Citation25].
Figure 11. Schematic representation of the S2 and S1 potential energy surfaces in the vicinity of the S2/S1 C2v CI. Colour indicates the electronic character of each surface which changes at the CI: green corresponds to the B2 and red to the A1 character. Arrows represent the branching of the system's trajectories/nuclear wavefunction in the approach to the CI. In this representation, the y-direction is the totally symmetric BLA mode represented in (a) and (b), which corresponds to the gradient and the S2/S1 gradient difference direction for the system approaching the CI. The x-direction is the non-totally symmetric in-plane distortion mode in (c) and (d), corresponding to the non-adiabatic derivative coupling direction for the system approaching the CI.
A second reason for deviations from the initial mass-weighted steepest descent path, and more important from the photochemistry point of view, is that along the initial sections of this path on the 1B2 surface both molecules are unstable with respect to the disrotatory motion of the butadiene sub-unit. We therefore initiated a second reaction path, represented as dashed lines in and , starting close to the S2/S1 CI but with a small disrotatory distortion. This path will develop in an area of the S1 potential energy surface which has mostly a covalent character.
In the first stages of the disrotatory distorted reaction path of BD, the system largely mirrors the C2v path, with a predominance of the BLA distortion and no further disrotatory deformation until the system reaches a fully distended structure. For CPD, however, the BLA motion is accompanied by disrotatory motion (see ). This difference between the two systems can also be observed in the S2/S1 minimum energy conical intersection (MECI) structures shown in Figure . While the S2/S1 MECI for BD is almost planar (with a somewhat more extended structure than the one reported in the supplementary material of Ref. [Citation19]), with a small 5° disrotatory distortion, the CPD S2/S1 MECI is lower in energy with respect to the FC region and has a much bigger disrotatory distortion, similar to the one obtained in [Citation25].
This somewhat counter-intuitive comparatively lower propensity of the BD S1 excited state to deform out of plane could justify why a marked change in electronic character of the S1 state along the photochemical reaction path can in principle be observed in this system (for the trans-butadiene case, see [Citation58]), while being much more nuanced in CPD [Citation25]. The change in electronic character of the state is associated with a planar structure, when the symmetry labels B2 and A1 for the electronic state wavefunction are well defined, and the CI represented in the energy profile in or a closely lying avoided crossing. Given the propensity of CPD to deform out of planarity, the molecule evolves to a so-called ‘mixed state’ quickly after excitation, when it becomes difficult to assign an ionic or covalent character to the S1 electronic state [Citation25].
After reaching the extended, almost planar structure, the reaction path of BD in starts a more pronounced disrotatory deformation, accompanied by a slight regression on the BLA motion, until it reaches a local minimum on the S1 surface with Cs symmetry. This is in agreement with previous reports [Citation15,Citation16,Citation59].
Photochemistry of BD is thought to further proceed by a backbone dihedral distortion, which is likely to occur with a small energy barrier [Citation15–17,Citation20,Citation60], until a CI between S1 and S0 with a so-called cisoid structure is reached. We thus proceeded to search for the lowest energy points along the S1/S0 seam using the RASSCF methodology. For BD, the MECI between the two lowest states had been optimised at a CASSCF(4,4) level [Citation20]. Starting from this geometry, we build a RASSCF(3,4,6)+3p active space from NBOs, as described in Sections 2 and 4.1. The result of this optimisation is the MECI structure 1.80 eV (42 kcal mol− 1) below the FC region displayed in Figure . The result is qualitatively similar to the starting structure, but with one-third of the molecule staying roughly planar and a terminal carbon distorting out of plane with a dihedral angle in the carbon backbone of 57.4°.
In contrast with the reaction path of BD, the reaction path of CPD does not reach a local minimum on the S1 surface. After about 20° of disrotatory distortion (see ), the molecule starts a more pronounced butadiene unit dihedral deformation which breaks the symmetry of the molecule and eventually leads to an S1/S0 CI which largely resembles the MECI structure reported in Figure (f). This shows that a barrierless photochemical reaction path exists between the FC region and an S1/S0 CI for CPD at our chosen RASSCF level of theory, in agreement with Ref. [Citation25].
Ref. [Citation25] reports finding three MECIs between the S1/S0 in CPD calculated at the MS-CASPT2 level with a CASSCF(4,4) reference wavefunction. Two of these result from torsion around one of the two double bonds, whereas the last one results from a disrotatory motion where both double bonds in CPD are twisted. All these structures were reported to lie in a very narrow interval of energies, suggesting a very flat intersection space landscape. For each of these structures, we constructed a RASSCF(5,4,5)+3p active space from NBOs, and have obtained in all three cases an energy gap between S1 and S0 of more than 1 eV. We note, however, that the potential energy surfaces are particularly steep in this region of nuclear coordinates (see the S0 surface in ), and reoptimising each of these structures, all three calculations converged to a single MECI structure shown in Figure . The optimised structure has four carbon atoms (including that of the CH2 group) roughly in a plane, with the remaining carbon atom distorting out of plane in an analogous arrangement to the BD MECI. Although different, the obtained structure most resembles structure eth1 of Ref. [Citation25], the lowest in energy from the three originally reported, via which more than 70% of the excited-state population decays to the ground state [Citation25] and closest to the structure at the end of the reaction path we calculated.
5. Concluding remarks
The simultaneous description of ionic and covalent electronic excited states still presents a challenge to current electronic structure methods. This is particularly important in studying photochemistry, when the electronic character of the molecular system changes along the reaction path and a balanced description of ionic and covalent states critically affects the shape of the potential energy surfaces.
In this study, we have illustrated for two simple, but challenging, cis-diene systems how RASSCF is able to provide a qualitatively correct description of the electronic structure of the three lowest valence states in the FC region, but also how it gives a correct account of the photochemical path of these systems, with relatively modest computational requirements.
We have shown how a RASSCF active space can be constructed from NBOs, with the orbital choice being guided by the concept of correlating orbital pairs/oscillator orbitals and the order of the configuration interaction expansion guided by the concepts of semi-internal and dynamical correlation.
For cis-butadiene, both semi-internal correlation between the π system and the σ framework and dynamical correlation of π orbitals provide important contributions to the stabilisation of the ionic excited state. For cyclopentadiene, σπ semi-internal correlation effects dominate over π dynamical correlation.
Acknowledgements
J.P. Malhado acknowledges the research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007-2013) under REA grant agreement 302639 EnPhoC. Calculations were performed at the Imperial College London High Performance Computing Centre.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed at http://dx.doi.org/10.1080/00268976.2015.1025880.
Additional information
Funding
Notes
1. Initially, one might be tempted to suggest that the single excitations of semi-internal correlation are merely polarisation effects that are usually included in the orbital optimization. However, as discussed by Sinanoğlu and others, this is true correlation between electrons in the RAS1 space and electrons in the RAS2 space that cannot be recovered as a result of orbital optimization.
2. This can be achieved using the forceabeliansymmetry keyword in the Gaussian program.
3. This is achieved using the nocpmcscf keyword in the Gaussian program.
References
- F. Bernardi, M. Olivucci, and M.A. Robb, Chem. Soc. Rev. 25(5), 321–328 (1996).
- M. Garavelli, Theor. Chem. Acc. 116, 87 (2006).
- L. Gonzalez, D. Escudero, and L. Serrano-Andrés, Chem. Phys. Chem. 13, 28 (2012).
- L. Blancafort, Chem. Phys. Chem. 15, 3166 (2014).
- J.M.O. Matos, B.O. Roos, and P.-Å. Malmqvist, J. Chem. Phys. 86, 1458 (1987).
- L. Serrano-Andrés, M. Merchán, I. Nebot-Gil, B.O. Roos, and M. Fülscher, J. Am. Chem. Soc. 115(14), 6184–6797 (1993).
- L. Serrano-Andrés, B.O. Roos, and M. Merchán, Theor. Chim. Acta 87(4–5), 387–402 (1994).
- N. Kuritz, T. Stein, R. Baer, and L. Kronik, J. Chem. Theory Comput. 7, 2408 (2011).
- B.J. Levine, C. Ko, J. Quenneville, and T.J. Martínez, Mol. Phys. 104(5–7), 1039–1051 (2006).
- J. Olsen, B.O. Roos, P. Jørgensen, and H.J. Aa. Jensen, J. Chem. Phys. 89(4), 2185–2192 (1988).
- P.-Å. Malmqvist, A. Rendell, and B.O. Roos, J. Phys. Chem. 94(14), 5477–5482 (1990).
- M. Klene, M.A. Robb, L. Blancafort, and M.J. Frisch, J. Chem. Phys. 119, 713 (2003).
- S. Vancoillie, H. Zhao, V.T. Tran, M.F.A. Hendricks, and K. Pierloot, J. Chem. Theory Comput. 7, 3961 (2011).
- G. Tomasello, M. Garavelli, and G. Orlandi, Phys. Chem. Chem. Phys. 15(45), 19763–19773 (2013).
- M. Olivucci, I.N. Ragazos, F. Bernardi, and M.A. Robb, J. Am. Chem. Soc. 115(9), 3710–3721 (1993).
- P. Celani, F. Bernardi, M. Olivucci, and M.A. Robb, J. Chem. Phys. 102 (14), 5733–5742 (1995).
- M. Garavelli, B. Frabboni, M. Fato, P. Celani, F. Bernardi, M.A. Robb, and M. Olivucci, J. Am. Chem. Soc. 121(7), 1537–1545 (1999).
- M. Garavelli, F. Bernardo, M. Olivucci, M.J. Bearpark, S. Klein, and M.A. Robb, J. Phys. Chem. A 105(51), 11496–11504 (2001).
- M. Boggio-Pasqua, M.J. Bearpark, M. Klene, and M.A. Robb, J. Chem. Phys. 120(17), 7849–7860 (2004).
- F. Sicilia, L. Blancafort, M.J. Bearpark, and M.A. Robb, J. Chem. Theory Comput. 4(2), 257–266 (2008).
- Y. Harabuchi, S. Maeda, T. Taketsugu, N. Minezawa, and K. Morokuma, J. Chem. Theory Comput. 9, 4116 (2013).
- R.J. Cave and E.R. Davidson, J. Phys. Chem. 91(17), 4481–4490 (1987).
- W. Fuß, W.E. Schmid, and S.A. Trushin, Chem. Phys. 316(1–3), 225–243 (2005).
- O. Schalk, A.E. Boguslavskiy, and A. Stolow, J. Phys. Chem. A 114(12), 4058–4064 (2010).
- T.S. Kuhlman, W.J. Glover, T. Mori, K.B. Møller, and T.J. Martínez, Faraday Discuss. 157, 193 (2012).
- Y.J. Bomble, K.W. Sattelmeyer, J.F. Stanton, and J. Gauss, J. Chem. Phys. 121(11), 5236–5240 (2004).
- K. Fukui, Acc. Chem. Res. 14(12), 363–368 (1981).
- R.J. Buenker, S. Shih, and S.D. Peyerimhoff, Chem. Phys. Lett. 44(2), 385–393 (1976).
- J.M. Foster and S.F. Boys, Rev. Mod. Phys. 32(2), 300–302 (1960).
- S.F. Boys, Quantum Theory of Atoms, Molecules and the Solid State, edited by P.-O. Löwdin (Academic Press, New York, 1963), p. 253.
- O. Sinanoğlu, Adv. Chem. Phys. 6, 315 (1964).
- İ. Öksüz and O. Sinanoğlu, Phys. Rev. 181(1), 42–53 (1969).
- V. Veryazov, P.Å. Malmqvist, and B.O. Roos, Int. J. Quantum Chem. 111, 3329 (2011).
- F. Krausbeck, D. Mendive-Tapia, A.J.W. Thom, and M.J. Bearpark, Comp. Theor. Chem. 1040–1041, 14 (2014).
- B.J. Rosenberg, W.C. Ermler, and I. Shavitt, J. Chem. Phys. 65, 4072 (1976).
- J.P. Foster and F. Weinhold, J. Am. Chem. Soc. 102(24), 7211–7218 (1980).
- R. McWeeny, Rev. Mod. Phys. 32, 335 (1960).
- S. Rothenberg and E.R. Davidson, J. Chem. Phys. 45 (7), 2560–2576 (1966).
- J.J.W. McDouall, Computational Quantum Chemistry: Molecular Structure and Properties in Silico (Royal Society of Chemistry, Cambridge, 2013).
- P.-Å. Malmqvist and B.O. Roos, Theor. Chim. Acta 83, 191 (1992).
- W.T. Borden and E.R. Davidson, Acc. Chem. Res. 29(2), 67–75 (1996).
- C. Angeli, J. Comput. Chem. 30(8), 1319–1333 (2009).
- M.A.C. Nascimento and W.A. Goddard, III,Chem. Phys. 36(2), 147–160 (1979).
- K. Pierloot, Computational Organometallic Chemistry (CRC Press, London, 2001), p. 123.
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, Jr, J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudinand, V.N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, P.V. Parandekar, N.J. Mayhall, A.D. Daniels, O. Farkas, J.B. Foresman, J.V. Ortiz, and J. Ciosl, Gaussian Development Version, Revision h.32 (Gaussian Inc., Wallingford, CT, 2010).
- K.A. Nguyen and M.S. Gordon, J. Am. Chem. Soc. 117(13), 3835–3847 (1995).
- D. Feller and N.C. Craig, J. Phys. Chem. A 113(8), 1601–1607 (2009).
- J. Almlöf and P.R. Taylor, Int. J. Quantum Chem. 27, 743 (1985).
- H.P. Hratchian and H.B. Schlegel, J. Phys. Chem. A 106(1), 165–169 (2002).
- M.E. Squillacote, R.S. Sheridan, O.L. Chapman, and F.A.L. Anet, J. Am. Chem. Soc. 101(13), 3657–3659 (1979).
- Stephen R. Langhoff, editor,Quantum Mechanical Electronic Structure Calculations with Chemical Accuracy (Springer Netherlands, Dordrecht, 1995).
- J.D. Watts, S.R. Gwaltney, and R.J. Bartlett, J. Chem. Phys. 105 (16), 6979–6988 (1996).
- M. Schreiber, M.R. Silva-Junior, S.P.A. Sauer, and W. Thiel, J. Chem. Phys. 128(13), 134110 (2008).
- S.P.A. Sauer, M. Schreiber, M.R. Silva-Junior, and W. Thiel, J. Chem. Theory Comput. 5(3), 555–564 (2009).
- Q. Shang and B.S. Hudson, Chem. Phys. Lett. 183(1–2), 63–68 (1991).
- L. Salem, Electrons in Chemical Reactions (Wiley, New York, 1982).
- J.P. Malhado, M.J. Bearpark, and J.T. Hynes, Front. Chem. 2, 97 (2014).
- B.G. Levine and T.J. Martínez, J. Phys. Chem. A 113(46), 12815–12824 (2009).
- S. Sakai, Chem. Phys. Lett. 287, 263 (1998).
- F. Sicilia, L. Blancafort, M.J. Bearpark, and M.A. Robb, J. Phys. Chem. A 111(11), 2182–2192 (2007).