

ABSTRACT
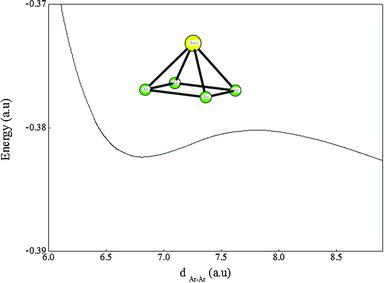
This study is interested in the illustration of ab initio potential energy curves for Ba+Arn (n = 1–4) clusters. The electronic structures of these molecules are calculated using [Ba2+] and [Ar] non-empirical core pseudo-potentials complemented by the core polarisation operators for both atoms, which allow the consideration of core valence correlation effects. The structure and stabilities of Ba+Arn (n = 1–4) clusters are investigated. These molecules are treated as one-electron active system. Spectroscopic constants and vibrational energy levels have been derived from their potentials. The analysis of the geometric forms, basing on the potential energy curves and the spectroscopic constants, clearly shows the importance of rare gas induced dipole. We also show that the dipolar interactions can influence the coupling between atoms.
Acknowledgements
We thank Miss Kawther Abdessalem, PhD in Quantum Physics Laboratory, University of Monastir, for helpful discussion.