
ABSTRACT
Chemical bonding in the highly selective hydrogenation catalyst GaPd is analysed by means of quantum chemical calculations employing the bonding analysis techniques in real space, in particular the quantum theory of atoms in molecules, the delocalisation indices and the electron localisability approach. A three-dimensional system of predominantly two-centre electron-deficient Ga–Pd interactions is revealed, being responsible for the high stability of the compound under hydrogenation conditions.
GRAPHICAL ABSTRACT
Introduction
The intermetallic compound PdGa was first discovered in 1947 [Citation1]. Taking into account the charge transfer found previously [Citation2] and clearly confirmed in this work, we hereafter call this compound as GaPd, i.e. gallium palladide. The crystal structure of the FeSi type (space group P213, Pearson symbol cP8, a = 4.880 Å) was suggested for GaPd on the basis of X-ray powder diffraction data [Citation1]. Further studies in the binary Ga–Pd and related ternary systems confirmed the structure-type assignment giving markedly different lattice parameters of a = 4.890 Å [Citation3,Citation4] and 4.965 Å [Citation5]. Just recently, the crystal structure was re-refined using X-ray single-crystal diffraction data yielding an intermediate value of a = 4.89695(6) Å [Citation6].
For a long time, GaPd was known solely as one of the representatives of the relatively simple (eight atoms in the unit cell) but non-centrosymmetric cubic structure motif. Later it was found to play a role in electronics, being formed by applying Al–Pd or Au–Pd–Pt alloys as Schottky contacts on GaAs to create metal semiconductor field effect [Citation7] and heterojunction bipolar transistors [Citation8]. Recently, GaPd was discovered to be an excellent catalyst for the industrially important heterogeneous semi-hydrogenation of acetylene to ethylene – even in the presence of a high excess of ethylene [Citation2,Citation9–12]. In catalysis, GaPd possesses very high structural stability and does not form hydrides in contrast to elemental palladium [Citation13]. The reason for the stability of GaPd under catalytic conditions or preferable formation of GaPd on the contact interfaces in electronic devices may lie in dedicated atomic interactions (chemical bonding) in the compound.
Studies concerning the chemical bonding in intermetallic compounds with the FeSi type of structure are rare, despite the fact that this structure type is realised by more than 60 intermetallic phases. The first interpretation of the short MSi interatomic distances in the prototype FeSi and the isotypic MSi compounds (M=Cr–Ni) was given by Pauling and Soldate [Citation14], who discussed the sevenfold coordination within the resonating-valence-bond theory [Citation15]. Following this analysis, Fe–Si as well as Fe–Fe interactions are present in the compound, the latter leading to the preference of the FeSi type above the NaCl type of structure. A study of possible structure-governing factors and an attempt to explain the atomic interactions in the FeSi type of structure by the Ortskorrelation (i.e. assuming a kind of spatial ordering within the electron gas) was made in [Citation16–18]. Neither the heteroatomic coordination by seven ligands for each atom, nor the formation of molecular units [Citation19,Citation20] could explain the formation of chemically different compounds crystallising in the FeSi type. On the other hand, the electron concentration indicates that optimised covalent bonding might play a role for the realisation of this type of crystal structure [Citation18]. Finally, a complex explanation for the occurrence of the FeSi type of structure was proposed using the multiple Ortskorrelation model [Citation21]. The presence of Ga–Pd bonds in GaPd was concluded applying X-ray absorption spectroscopy at the Pd L2,3 edges [Citation22]. According to the interpretation of these measurements, charge is transferred from Pd to Ga, being contradictory to the expectations from the difference in Pauling's or absolute electronegativity between the two elements [Citation23]. The FeSi type of crystal structure with its relatively low Madelung factor of 1.6839 (calculated for the idealised atomic parameters of x(Fe) = (√5 − 1)/8 ≈ 0.15451; x(Si) = −x(Fe)) is less predestined for ionic bonding, since the NaCl as well as the CsCl types of structure possess more favourable Madelung factors of 1.7476 and 1.7627, respectively [Citation24]. Besides these attempts to explain the realisation of the FeSi type of structure, a first bonding analysis in real space has been performed for GaPd on the basis of LMTO-ASA calculations revealing a system of two- and three-centre interactions within the crystal structure [Citation2]. Here, the results of the analysis of the bonding interactions in GaPd applying different combined quantum mechanical techniques in real space are reported.
Calculation techniques
The quantum chemical calculations were performed using the unit cell parameter and atomic coordinates from the most recent structure determination [Citation6]. Calculations with different methods were performed to analyse the different aspects of the atomic interactions as well as the stability of the results with respect to the calculation approach. Because mainly the real-space techniques were used for the analysis of chemical bonding in GaPd, they were compared on the level of calculated electron density (ED). The optimisation of structural parameters with LDA–Perdew–Wang or GGA–PBE potentials did not reveal a significant influence on the quality of the resulted ED (full-potential local orbital method (FPLO) calculation, cf. below) supporting the use of experimental structural parameters for calculations.
For the calculation using the TB-LMTO-ASA program package [Citation25], the Barth–Hedin exchange potential [Citation26] was employed for the LDA calculations. The calculation within the atomic sphere approximation (ASA) includes corrections for the neglect of interstitial regions and partial waves of higher order [Citation27], explicit addition of empty spheres was not necessary. The following radii of the atomic spheres were applied for the calculations for the GaPd compound: r(Pd) = 1.502 Å, r(Ga) = 1.535 Å. A basis set containing Pd(5s,5p,4d) and Ga(4s,4p) orbitals was employed for a self-consistent calculation with Pd(4f) and Ga(4d) functions being downfolded. Crystal Orbital Hamilton Populations (COHP) were calculated according to [Citation28] with a special module implemented into the TB-LMTO-ASA program package.
Further first-principles electronic structure calculations within the local density approximation were performed using version 9.01 of the all-electron FPLO code [Citation29]. Exchange-correlation effects were taken into account by employing the Perdew–Wang parameterisation [Citation30]. The valence (5s,6s,4d,5d,5p for Pd, 4s,5s,4p,5p,4d for Ga) and semi-core states (4s, 4p for Pd, 3s,3p,3d for Ga) were treated at the scalar-relativistic level, while a fully relativistic approach was employed for the lower-lying core states. The Brillouin zone was sampled by a well-converged grid of 24×24×24. The atom-centred charge densities were expanded up to lmax = 12. For the optimisation test, the LDA–Perdew–Wang [Citation30] and GGA–PBE [Citation31] potentials were used. The obtained structural parameters do not differ much as expected between each other and from the experimental data (GGA–PBE: a = 4.974 Å, x(Pd) = 0.392, x(Ga) = 0.093; LDA/Perdew–Wang: a = 4.843 Å, x(Pd) = 0.393, x(Ga) = 0.093; experiment: a = 4.89695 Å, x(Pd) = 0.3924, x(Ga) = 0.09295 [Citation6]).
The calculation of the localisation and two-centre delocalisation indices (LI/DI) for solids [Citation32,Citation33] as well of the new bonding indicator C0.6 [Citation34] and their subsequent analysis were performed from the results of full-potential scalar-relativistic (L)APW+lo+LO band structure calculations (Elk code [Citation35]) with the program DGrid [Citation36]. The three-centre DIs (principally defined in [Citation37]) were calculated between QTAIM (quantum theory of atoms in molecules) atoms according to [Citation38] with the program DISij [Citation39] using the atomic overlap matrices [Citation32] as computed by DGrid. For the Elk calculations, the Gkmax parameter was set to 9, the MT radii for Pd and Ga were equal to 2.0 a.u., and a mesh of 4×4×4 k points was used.
The quantum chemical program system ADF [Citation40] was used to calculate the DFT wavefunction (within LDA) for molecular fragments of the crystal structure of GaPd using triple-zeta basis sets (Slater functions) with two sets of polarisation functions (‘TZ2P’) for Pd and Ga.
The electron localisability indicator (ELI) in its ELI-D representation (ΥD) [Citation41,Citation42] was evaluated with modules implemented within the TB-LMTO-ASA [Citation25] and FPLO program packages [Citation43]. The topology of ELI-D and ED was analysed using the program DGrid [Citation36] with consecutive integration of the ED in basins, which are bound by zero-flux surfaces in the ELI-D or ED gradient fields. This procedure, based on that proposed by Bader for the ED [Citation43], allows to assign an electron population for each ELI-D or ED basin, yielding additional relevant information about the chemical bonding.
Results and discussion
Assuming that the basic coordination of atoms remains basically unchanged on the surface, the necessary feature to create isolated active atomic centres on the surface of a catalytic material is heteroatomic-only environment of the catalytically active atoms in the bulk crystal structure. There are few inorganic crystal structures revealing such a feature. For coordination number of four this is the structure of the sphalerite type, for coordination number of six this is the rock-salt type of structure. In the non-centrosymmetric structure type NbAs each atom has six heteroatomic with eight homoatomic ligands in the next sphere. The representatives of this type – in particular NbP [Citation44] – were very recently found to be good candidates for the so-called topological Weyl semimetals with a range of exotic transport properties and electronic surface states. This indicates that the heteroatomic coordination plays probably a basic role for surface behaviour of materials. In case of coordination number of seven, there is only one structure type – FeSi – known for the heteroatomic-only environment of atoms. In the structure of the α-TlI type each atom has seven heteroatomic and two homoatomic ligands.
The cubic crystal structure of GaPd belongs to the structure type FeSi and contains eight atoms per unit cell (, top). All shortest interatomic distances are heteroatomic. Based on the recent re-refinement of the crystal structure [Citation6] the shortest contact between Ga and Pd of d1 = 2.54 Å is along the three-fold axis (pink in ). The next contacts are slightly longer (d2 = 2.57 Å, blue in ). These distances are well comparable with the sum of the covalent radii of gallium (1.25 Å) and palladium (1.28 Å [Citation23]). The next longer contacts of d3 = 2.71 Å (yellow in , top) are already comparable with the atomic (metallic) radii of Pd (1.376 Å) and Ga (1.41 Å) [Citation23]. In addition to the shortest heteronuclear contacts, each atom has six longer homonuclear contacts of d(Ga–Ga) = 3.03 Å and d(Pd–Pd) = 3.01 Å. These are only about 10% larger than the sum of the metallic radii.
Figure 1. Crystal structure of GaPd: (top) unit cell with the three types of shortest Ga–Pd distances; (bottom) atomic environment of palladium (left) and gallium (right).
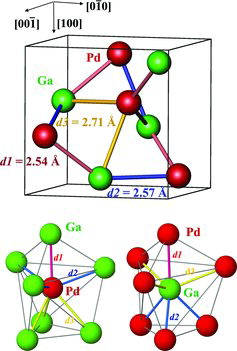
For each Ga or Pd atom, there is one heteronuclear ligand with the shortest distance, and 3 + 3 others with the two longer distances, yielding for both atoms the same (heteronuclear) coordination number of seven and very similar coordination polyhedrons with trigonal C3v symmetry (, bottom). Such distribution of the interatomic distances within a coordination sphere of an atom – shortest contacts close to the sum of covalent radii, longer distances comparable rather with the sum of atomic (metallic) radii, often also with a group of intermediate distances – is on one hand characteristic for many intermetallic compounds, e.g. for the chemically related binary compound Ga2Ir [Citation45]. On the other hand, it does not allow making ad hoc suggestions about the type of chemical bonding in the crystal structure using the interatomic distances, as usually possible for the valence inorganic and organic compounds.
The peculiar shape of the coordination polyhedrons in GaPd in combination with the relatively low nearest-neighbour coordination number of seven for both atom types and the short Ga–Pd distances suggest significant-directed (covalent) bonding, which is consistent with the brittleness of this substance. Of course, the latter may also be caused by ionic interactions which were earlier suggested in the Ga–Pd system already due to the electronegativity difference between gallium and palladium [Citation1]. Against this suggestion, the Madelung factor of the FeSi type of structure (1.6839, cf. Introduction section) is much lower than the corresponding values for the NaCl as well as the CsCl types of structure (1.7476 and 1.7627, respectively) suggesting rather reduced ionic contributions to the bonding in GaPd [Citation24]. An additional indication for the presence of strong bonding for GaPd is the rather high standard enthalpy of formation ΔfH0 = −143.2 ± 4.6 kJ/mol [Citation46].
The interatomic distances in GaPd can be interpreted calculating Pauling bond orders (PBO [Citation47]) applying covalent radii (see above) as single-bond radii. With this assumption, for the shorter d1 and d2 distances the obtained PBO value are close to unity: PBO(d1) = 0.96 and PBO(d2) = 0.84, respectively. For the longer distance d3, the PBO value drops to 0.51. The (heteroatomic) PBO sums within the first coordination sphere for both, Ga and Pd, are 5.01. While for a palladium atom this number is not surprising taking into account the nine valence orbitals available for bonding, it is hard to imagine how Ga can realise five bonds with seven neighbours employing four available valence orbitals. Thus, more thoughts on atomic interactions should be made.
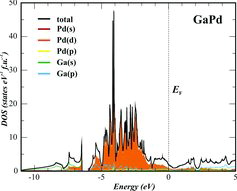
Further information about the character of bonding in a crystal structure can be extracted from the calculated electronic structure. The electronic density of states (DOS) for GaPd is minor dependent on the calculation techniques. The here presented DOS (), calculated with the FPLO code, shows-similarly to the previously published TB-LMTO-ASA calculations [Citation2]-one low-energy region (E < −6.5 eV) formed mainly by Ga(s) and Pd(d) states. It is clearly separated from a large region, which – in turn – can be partitioned into three sub-regions. The first one (−6 eV < E < −5 eV) is composed of comparable contributions of Ga(p) and Pd(d) states, the largest sub-region (−5 eV < E < −1.2 eV) is mainly formed by (most probably non-bounding) Pd(d) states with minor contributions of Ga(p) states. The sub-region just below the Fermi level is again built of comparable contributions of Pd(d) and Ga(p) states. The contributions of s states of palladium are distributed over wide energy range below the Fermi level. Splitting of the palladium d states and their energetic overlap with the s and p states of gallium support the – from the crystal structure analysis already indicated – bonding relevance of the heteronuclear Ga–Pd interactions.
Figure 2. Total electronic density of states (DOS) for GaPd together with partial atomic contributions (FPLO code).
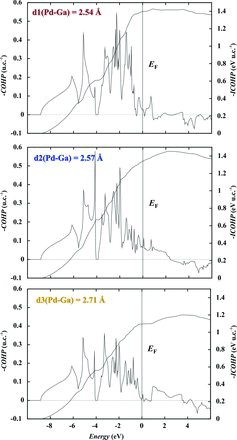
The stabilising character of the Ga–Pd interaction along d1, d2 and d3 contacts is clearly revealed by the COHP analysis (TB-LMTO-ASA calculation, ). Positive values of −COHP in the whole energy region below the Fermi level for all three contacts above reveal the bonding character of the Ga–Pd interactions along these contacts. Large values of 1.4 and 1.3 for the integrated COHP (ICOHP) at the Fermi level for d1 and d2, respectively, are well in agreement with the short interatomic distances. As expected, for the longer d3 contact the value of ICOHP(EF,d3) = 1.1 is markedly smaller. The behaviour of COHP(E) around EF in GaPd differs from the situation in compounds which can be described with classical valence rules or in Zintl phases. While in the latter groups the COHP(E) for bonding interactions is normally saturated at EF, for all three interactions d1, d2 and d3 in GaPd, positive contributions to –COHP(E) continue up to 4 eV above EF. The d1, d2 and d3 Ga–Pd interactions are caused by the same type of orbitals, namely the 5s and 4d states of Pd and 4s and 4p states of Ga.
Figure 3. COHP analysis of atomic interactions in GaPd (colour coding as in ).
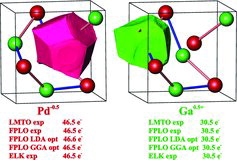
To shed more light on the ionic part of the atomic interactions, the effective charges of palladium and gallium species in GaPd were calculated applying the QTAIM technique [Citation48]. Topological analysis of the ED reveals very similar shapes of the QTAIM basins for Pd and Ga reflecting similar geometry of atomic environment, assuming the predominantly covalent interactions (). Practically independent of the calculation technique, the total population of 46.5 electrons was found for the Pd basin and 30.5 electrons for the Ga basins revealing a charge transfer of half an electron from Ga to Pd. This is in agreement with the electronegativities of the elements in the Pearson or absolute scale (ENPd > ENGa). On the one hand, the obtained relatively small charge transfer reveals moderate contribution of ionic interactions to the bonding picture in GaPd and thus correlates with the earlier expectations based on the analysis of Madelung factors [Citation24]; on the other hand it is markedly smaller than the transfer of 1.9 electrons from Al to Pt observed for the chemically related equiatomic compound AlPt (structure type CsCl) [Citation49] even considering the larger difference of electronegativities between aluminium and platinum if comparing with that between Ga and Pd, as well as the difference in the Madelung factors between the CsCl and PdGa types of structure. The observed moderate charge transfer rather points out to covalent polar Ga–Pd interactions instead of ionic ones.
Figure 4. QTAIM atoms (basins) and their charges (populations) in GaPd obtained from the calculations with different codes. The extension exp means that the experimental values of the atomic coordinates were used, opt means optimisation of the atomic coordinates applying LDA or GGA potentials.
This polar character of Ga–Pd interactions is supported by the analysis of the spatial distribution of the bonding indicator C0.6, a new quantum chemical tool based on the ED inhomogeneity [Citation34]. According to first studies [Citation50], polar bonds are doubly indicated, i.e. the indicator C0.6 exhibits two separate maxima in a bonding region, if the bond is sufficiently polar. Exactly such a distribution of C0.6 was found for GaPd () revealing the polar nature of the heteronuclear interactions. The attractor on the Pd side originates from the outermost atomic shell of Pd, and therefore indicates that Pd is the more electronegative partner in this bonding situation. Those findings are well in agreement with the conclusions of the QTAIM analysis.
Figure 5. Bonding indicator C0.6 in GaPd: (top) location of C0.6 maxima (black) in vicinity of the Ga–Pd contacts; (bottom) C0.6 distribution on the Pd plane (colour coding is analogous to one for ELI-D).
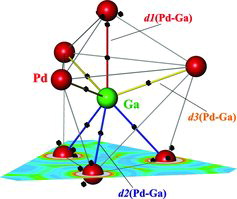
The nominal number of 2.5 valence electrons per atom in GaPd (using the characteristic valences two and three for Pd and Ga, respectively) is smaller than that in normal valence inorganic compounds and is in the region typical for Zintl phases [Citation51], cf. 2.5 e− for Na4Si4. Nevertheless, the bonding in GaPd, in particular seven heteroatomic short contacts per atom, cannot be understood in terms of the Zintl concept [Citation52,Citation53]. For further understanding of the spatial organisation of the atomic interactions in GaPd the electron localisability approach [Citation54] is applied. The electron localisability indicator ELI-D was calculated using the dedicated module within the FPLO program package [Citation43]. Analysis of the ELI-D distribution revealed three sets of maxima in the unit cell (, top):
| (i1) | attractor in the triangle Ga–Pd(d1)–Pd(d3), each three of them are located around the d1(Pd–Ga) contact; the basin of this attractor has a common surface with the three core basins mentioned above (i.e. it is tri-synaptic) and formally may represent a three-centre interaction; the population of this basin is 0.57 electrons, the atomic contributions are 0.34 from Ga, 0.10 from Pd(d1) and 0.13 from Pd(d3); | ||||
| (i2) | attractor in the triangle Ga-Pd(d2)-Pd(d3), there are three of them per Ga atom; this triangle is formed by d2 and d3 as well as the long d(Pd–Pd) contact of 3.05 Å; on the basis of its tri-synapticity this attractor may also represent a three-centre bonding; the population of this basin is 0.63 electrons, the atomic contributions are 0.35 from Ga, 0.15 from Pd(d2) and 0.13 from Pd(d3); | ||||
| (i3) | attractor located at the gallium atom on the bond-opposite side of d1 contact within the tetrahedron formed by one Ga and three Pd(d2) atoms; topologically it is tetra-synaptic one and may represent a four-centre interaction; this basin is populated with 1.22 electrons, the atomic contributions are 0.68 from Ga and 0.18 from each Pd. | ||||
Figure 6. Distribution of ELI-D for triplet-coupled electrons [Citation55] in GaPd crystal and related molecular species GaPd (triplet state), GaPdH (singlet state) and PdGaPd3 (triplet state).
![Figure 6. Distribution of ELI-D for triplet-coupled electrons [Citation55] in GaPd crystal and related molecular species GaPd (triplet state), GaPdH (singlet state) and PdGaPd3 (triplet state).](/cms/asset/66466380-978f-4cf7-99ae-99d5eb74c6d8/tmph_a_1093664_f0006_oc.jpg)
Counting the contributions of each atom to the bonding basins in its vicinity yields 2.75 (less than 3) electrons for Ga (3×0.34 + 3×0.35 + 1×0.68) and 2.28 (more than 2) electrons for Pd (3×0.10 + 6×0.15 + 6×0.18). These values are close to the numbers suggested above by the summed PBO = 5.01 (requiring two electrons from Pd and three from Ga). Furthermore, they support the direction of the charge transfer obtained from the QTAIM analysis. This result also goes in line with the only light structuring of the penultimate shell of the palladium atom, i.e. deviation from the spherical distribution which is characteristic for the participation of the inner-shell electrons in the bonding within the valence region [Citation56,Citation57] and quantified by structuring index ϵ = 0.02 [Citation57]. Reduced values of ELI-D within penultimate the shell are located close to the d1, d2 and d3 contacts visualising the participation of the electrons of this shell in the bonding.
Looking on the topology of the ELI-D distribution in GaPd from the point of view of the synapticity of the ELI-D basins (see above), the atomic interactions may be interpreted as multi-centre (mostly three-centre) ones involving both palladium and gallium atoms. However, the direct-space bonding analysis of the interactions of transition metals, e.g. in the molecular species ScGa, Sc2 [Citation56] or Sc22+ [Citation56,Citation57], revealed that topologically these two-centre interactions are manifested not only by appearance of ring attractors between the cores, but also by structuring of the penultimate shells of the transition metals, as well as with attractors on the outer (bond-opposite) side of the participating atoms.
Thus, in order to understand more in detail chemical implications of each ELI-D attractor, ED and triplet ELI-D were calculated for the molecular species GaPd with the interatomic distance of d(Ga–Pd) = 2.486 Å (ADF code; ). Integration of ED revealed a charge transfer according to Ga+0.4Pd−0.4 which has the same direction but is slightly smaller than in the GaPd crystal due to the lower heteroatomic coordination number (one in the molecular species and seven in the crystal). An ELI-D attractor on the bond-opposite side visualising the fifth shell of the palladium atom is not observed, similar to the isolated atom [Citation58,Citation59]. The ELI-D distribution in the penultimate (fourth) shell deviates from the spherical one (structuring, cf. above) as expected for an interacting atom. Between the core regions, a ring attractor is found equivalent to i1 in the GaPd crystal. In addition to that, an attractor is found on the bond-opposite side of the gallium nucleus (lone-pair like, equivalent to i3 in the GaPd crystal). The population of the bond-ring-attractor basin is 1.8 electrons; the Ga lone-pair basin is populated with 2.0 electrons. In total 3.8 electrons participate in the bonding interactions within the GaPd molecule, which is assumed to be necessary amount for the two-centre only Ga–Pd interaction. Provided that all three valence electrons of gallium (3.2 for Hartree-Fock, non-relativistic approximation [Citation60] or 3.4 in DFT, scalar relativistic approximation [Citation59]) are used for bonding, a participation of only 0.8 (0.6 or even 0.4) electrons, respectively, is required from palladium.
Despite strong changes in the chemical composition, the characteristic topological features of the ELI-D distribution for a Ga–Pd bond – ring attractor between the atoms and second attractor on the bond-outer side at gallium – are sustainable in case of the linear molecule GaPdH with the hydrogen atom connected to gallium (d(Pd–Ga) = 2.49 Å, d(Ga–H) = 1.63 Å; ). The QTAIM charge of gallium (Ga+0.6Pd−0.2H−0.4) is larger as in 6aPd being in agreement with the appearance of the additional to Ga–Pd polar bond Ga–H. Here, the former bond-opposite attractor at the gallium nucleus (lone-pair, equivalent to i3) now represents the Ga–H bond. The basin populations of the Ga–Pd bond (ring) attractor i1 (2.6 e−) and the Ga–H bond attractor (2.1 e−) yield together 4.7 electrons, reflecting the larger amount of electrons in the system if compared with GaPd moiety. The additional electron is populating i1 attractor of the Ga–Pd bond. The contribution of palladium to the bonding basin is 0.7 (0.5 or even 0.3, cf. conditions for GaPd molecule) electrons being similar to that in the GaPd molecule.
Regardless of further changes in the chemical composition of the molecular species, the characteristic features of the ELI-D topology of the GaPd molecule (cf. above) are still present in the PdGaPd3 molecule with the interatomic distances and angles mimicking the bulk structure of GaPd (d1(Ga–Pd) = 2.486 Å, 3×d2(Ga–Pd) = 2.515 Å; ). Moreover, the distribution of ELI-D in PdGaPd3 shows also similarities with the picture in the bulk crystal structure. Because the symmetry of the molecule is no more cylindrical (as this was in case of Ga–Pd and Pd–Ga–H) but trigonal C3v, the ring attractor between the Pd and Ga cores with the short distance (d1, pink in ) is split into three with the population of 0.70 electrons each (like the i1 attractors in GaPd crystal, cf. also similar population). Also here, an ELI-D attractor is located on the bond-opposite side of the Ga atom within the tetrahedron GaPd3 (like the i3 attractors in GaPd crystal, population 0.23 electrons). The next attractors in the PdGaPd3 molecules are located in the i2 position in respect to the short Ga–Pd contacts and have the population of 0.67 electrons. Applying a gedanken-intersection between the basin of i3 attractor of the Ga lone-pair (cf. GaPd molecule) with the three basins of the i1 Ga–Pd interactions at the longer distance (blue in , bottom), the i3 attractor in PdGaPd3 may be interpreted as representing 4 two-centre Ga–Pd interactions rather than as an independent one revealing only the four-centre interaction. Following this gedanken-intersection scheme, the whole ELI-D distribution in PdGaPd3 may be considered as a ‘superposition’ of ELI-D distributions of 4 two-centre Ga–Pd interactions. So, each of the three basins of split ring attractors (i1 type) for the short Ga–Pd contact (pink, in ) is gedanken-intersecting with the basin of the bond-opposite attractor (i3 type) for the longer Ga–Pd contact (blue in ) and forms the basin of the i1 attractor in PdGaPd3. The remaining split i1 attractors' basins of the longer Ga–Pd contact (blue in , bottom) form by gedanken-intersection the basins of the i2 attractors in the molecule PdGaPd3. This happens despite the fact that the complete heteroatomic environment of Ga in this hypothetic molecule is not finished (only four of the seven Pd atoms are present). The total amount of electrons in the environment of the Ga atom is 4.34 being closer to 4.82 in the crystal structure of GaPd. Such ‘sharing’ or ‘superposition’ of the attractors' basins may be understood as a consequence of the insufficient number of electrons in the system to realise each Ga–Pd interaction with 3.8 electrons per formula unit (cf. GaPd molecule above).
In this way, the whole crystal structure of GaPd can be considered as a system of ‘two-centre’ Ga–Pd interactions with ‘shared’ attractors' basins in the ELI-D distribution. So – in addition to the described above – the basins of split ring attractors for the d1 and d2 interactions share basins with the split ring attractors for the d3 interaction (yellow in , top). The bond-opposite attractor for the d3 interaction is less pronounced and is shared with the ring attractors of the d2 interaction (, top). The populations of the corresponding ELI-D basins support this interpretation. In particular the populations of the split basins i1 and i2 in GaPd crystal are close to 1/3 of the population of the ring attractor in the GaPd molecule and close to the population of 0.65 electrons for the split ring attractors for the short Ga–Pd contact in PdGaPd3, being in agreement with the splitting scheme described above. The basin population of the i3 attractor in GaPd crystal (1.22 electrons) is noticeably larger as that of the bond-opposite attractor in the PdGaPd3 molecule (0.23 electrons).
The above suggested system of the two-centre electron-deficient Ga–Pd interactions is well in agreement with the calculated delocalisation indices. Of the total population of 46.5 e− for the QTAIM basin of the Pd anion (Elk code), 44.0 are localised and 2.5 are available for sharing (the variance σ2 of the electronic population for the QTAIM basin of Pd). For the Ga cation, 28.7 electrons of the total 30.5 in the QTAIM basin are localised and 1.8 electrons are available for interactions. The largest delocalisation indexes δ (real-space variant to the covalent bond order) are found for the two-centre contacts d1, d2 and d3: 0.41, 0.40 and 0.31, respectively. Furthermore, the next large delocalisation indices of δ = 0.26 and δ = 0.09 were found for the contact d(Pd–Pd) = 3.01 Å and d(Ga–Ga) = 3.03 Å. To quantify the three-centre contributions to the relevant interactions in the GaPd crystal, the three-centre bond delocalisation ratios G(d1) = 0.47, G(d2) = 0.45, G(d3) = 0.56, G(Pd,Pd) = 0.44 and G(Ga,Ga) = 1.75 were calculated [Citation39]. For the ideal three-centre-two-electron bond, the G value is 1. If G for a bond is much larger than 1 and the corresponding DI is very small, the bonding between the participating atoms can be neglected. This is the case for the Ga–Ga interaction. In contrast, the DI value δ(Pd,Pd) = 0.27 and its moderate delocalisation ratio G(Pd,Pd) = 0.44 indicate, that Pd tends to extend its relevant coordination by six homoatomic neighbours. This is consistent with the expected higher bonding capability of palladium compared to gallium, and – remarkably – has been predicted already by Pauling and Soldate [Citation14]. Thus, the values of G around 0.5 obtained for the Ga–Pd interactions reflect noticeable but small three-centre contributions to these bonds which are consistent with the interpretation of the topology of ELI-D above.
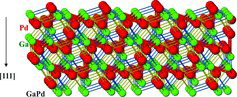
Assuming further that the directed Ga–Pd bonds play the major role for structural stability one can search in the crystal structure for the planes (surfaces) running mostly thorough the regions of Ga–Pd interactions. Such planes are located perpendicular to the [111] direction. With respect to the homogeneous distribution and density of the Ga–Pd interactions, the atomic decoration of a most promising plane is shown in . Despite several assumptions leading to the selection on this plane, the relative stability of the atomic distribution on the so-terminating surface of [111]-oriented GaPd single crystal was confirmed by scanning transmission microscopy experiments under the ultra high-vacuum conditions [Citation61].
Figure 7. Atomic decoration of the (-1-1-1) plane in the GaPd crystal structure with the highest accommodation of the d1 and d2 Ga–Pd interactions.
Conclusions
In the intermetallic compound GaPd, the number of available valence electrons (2.5 per atom if using the usual valences) is much smaller in comparison with inorganic valence compounds. It is rather similar to the electron counts in the Zintl phases. Because of a rather small charge transfer (QTAIM charges Ga0.5+ and Pd0.5−), the formation of the separated cations and Zintl anions is not possible which hinders interpretation of chemical bonding within the Zintl concept. All Ga–Pd interactions were found to be polar covalent. Chemical bonding in the crystal of GaPd is described as a system of heteroatomic two-centre electron-deficient Ga–Pd interactions. To realise all of them with the available electron count, ‘sharing’ the attractors' basins in the real-space representation is found as the suitable mechanism. The plane with the highest density of the Ga–Pd interactions was selected as the possible terminating one in the [111] direction in the GaPd crystal. This selection is well in agreement with the experimental data.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- E. Hellner and F. Laves, Z. Naturforsch. A 2, 177 (1947).
- K. Kovnir, M. Armbrüster, D. Teschner, T.V. Venkov, F.C. Jentoft, A. Knop-Gericke, Yu. Grin, and R. Schlögl, Sci. Techn. Adv. Mat. 8, 420 (2007).
- K. Khalaff and K. Schubert, J. Less-Common Met. 37, 129 (1974).
- M.K. Bhargava, A.A. Gadalla, and K. Schubert, J. Less-Common Met. 42, 69 (1975).
- S. Bhan and H. Kudielka, Z. Metallkd. 69, 333 (1978).
- M. Armbrüster, H. Borrmann, M. Wedel, Y. Prots, R. Giedigkeit, and P. Gille, Z. Kristallogr. NCS 225, 617 (2010).
- T.S. Huang and J.G. Pang, Mater. Sci. Eng. B 49, 144 (1997).
- D.G. Ivey, R. Zhang, Z. Abid, S. Eicher, and T.P. Lester, J. Mat. Sci. Mat. Electron. 8, 281 (1997).
- J. Osswald, R. Giedigkeit, R.E. Jentoft, M. Armbrüster, F. Girgsdies, K. Kovnir, T. Ressler, Yu. Grin, and R. Schlögl, J. Catal. 258, 210 (2008).
- J. Osswald, K. Kovnir, M. Armbrüster, R. Giedigkeit, R.E. Jentoft, U. Wild, Yu. Grin, and R. Schlögl, J. Catal. 258, 219 (2008).
- M. Armbrüster, K. Kovnir, M. Behrens, D. Teschner, Yu. Grin, and R. Schlögl, J. Am. Chem. Soc. 132, 14145 (2010).
- J. Osswald, R. Giedigkeit, M. Armbrüster, K. Kovnir, R.E. Jentoft, T. Ressler, Yu. Grin, and R. Schlögl, Patent EP1834939 (2006); WO2007104569 (2007).
- L.L. Jewell and B.H. Davis, Appl. Catal. A 310, 1 (2006).
- L. Pauling and A.M. Soldate, Acta Crystallogr. 1, 212 (1948).
- L. Pauling, Nature 161, 1019 (1948).
- K. Schubert, Z. Naturforsch. A 5, 345 (1950).
- K. Schubert and P. Eßlinger, Z. Metallkd. 48, 193 (1957).
- P. Eßlinger and K. Schubert, Z. Metallkd. 48, 126 (1957).
- F. Wever and H. Möller, Z. Kristallogr. 75, 362 (1930).
- C. Herman, O. Lohrmann, and H. Philipp, Strukturbericht 2, 13 (1937).
- K. Schubert, Z. Kristallogr, 179, 187 (1987).
- Y. Jeon, J. Chen, and M. Croft, Phys. Rev. B 50, 6555 (1994).
- J. Emsley, The Elements (Clarendon Press, Oxford, 1991).
- R. Fischer and J. Zemann, Monatsh. Chem. 103, 1613 (1972).
- O. Jepsen, A. Burkhardt, and O.K. Andersen, The Program TB-LMTO-ASA, version 4.7 (Max-Planck-Institut für Festkörperforschung, Stuttgart, 1999).
- U. Barth and L. Hedin, J. Phys. C 5, 1629 (1972).
- O.K. Andersen, Phys. Rev. B 12, 3060 (1975).
- R. Dronskowski and P.E. Blochl, J. Phys. Chem. 97, 8617 (1993).
- K. Koepernik and H. Eschrig, Phys. Rev. B 59, 1743 (1999).
- J.P. Perdew and Y. Wang, Phys. Rev. B 45, 13244 (1992).
- J.P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996).
- A. Baranov and M. Kohout, J. Comput. Chem. 32, 2064 (2011).
- A. Baranov, M. Kohout, and R. Ponec, J. Chem. Phys. 137, 214109 (2012).
- K. Wagner and M. Kohout, Theor. Chem. Acc. 128, 39 (2011).
- K. Dewhupt, S. Sharma, L. Nordström, F. Criccio, F. Bultmerk, O. Grånäs, and E. K.U. Gross, Computer code Elk 1.4.22. http://elk.sourceforge.net.
- M. Kohout, Program DGrid, version 4.6 (Radebeul, Germany, 2011).
- R. Ponec and F. Uhlik, Croat. Chem. Acta 69, 941 (1996).
- C. Börrnert, Yu. Grin, and F.R. Wagner, Z. Anorg. Allg. Chem. 639, 2013 (2013).
- F.R. Wagner, Program DISij, version 6.9.7 (Dresden, Germany, 2014).
- Computer code ADF2002.03 (SCM, Theoretical Chemistry, Vrije University, Amsterdam), http://www.scm.com.
- M. Kohout, Int. J. Quant. Chem. 97, 651 (2004).
- M. Kohout, Faraday Discuss. 135, 43 (2007).
- A. Ormeci, H. Rosner, F.R. Wagner, M. Kohout, and Yu. Grin, J. Phys. Chem. A 110, 1100 (2006).
- C. Shekhar, A.K. Nayak, Y. Sun, M. Schmidt, M. Nicklas, I. Leermakers, U. Zeitler, Y. Skourski, J. Wosnitza, Z.-K. Liu, Y.-L. Chen, W. Schnelle, H. Borrmann, Yu. Grin, C. Felser, and B.-H. Yan, Nature Phys. 11, 645 (2015).
- M. Boström, Yu. Prots, and Yu. Grin, Solid State Sci. 6, 499 (2004).
- S.V. Meschel and O.J. Kleppa, Thermochim. Acta. 292, 13 (1997).
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals, 3rd ed. (Cornell University Press, Ithaca, NY, 1960).
- R.F.W. Bader, Atoms in Molecules: A Quantum Theory (Oxford University Press, Oxford, 1999).
- A. Baranov, M. Kohout, F.R. Wagner, Yu. Grin, and W. Bronger, Z. Kristallogr. 222, 527 (2007).
- K. Finzel, Yu. Grin, and M. Kohout, Theor. Chem. Acc. 108, 1106 (2012).
- Yu. Grin, in Comprehensive Inorganic Chemistry II, edited by J. Reedijk and K. Poeppelmeier (Elsevier, Oxford, 2013), Vol. 2, p. 359.
- E. Zintl and W. Dullenkopf, Z. Phys. Chem. B 16, 195 (1932).
- R. Kniep, in Chemistry, Structure and Bonding of Zintl Phases and Ions, edited by S. Kauzlarich (VCH Publishers, New York, NY, 1996), p. xvii.
- M. Kohout, F.R. Wagner, and Yu. Grin, Int. J. Quant. Chem. 106, 1499 (2006).
- M. Kohout, F.R. Wagner, and Yu. Grin, Theor. Chem. Acc. 119, 413 (2008).
- M. Kohout, F.R. Wagner, and Yu. Grin, Theor. Chem. Acc. 108, 150 (2002).
- F.R. Wagner, V. Bezugly, M. Kohout, and Yu. Grin, Chem. Eur. J. 13, 5724 (2007).
- M. Kohout, Int. J. Quant. Chem. 83, 324 (2001).
- A.I. Baranov, J. Comp. Chem. 35, 565 (2014).
- M. Kohout and A. Savin, Int. J. Quant. Chem. 60, 875 (1996).
- D. Rosenthal, R. Widmer, R. Wagner, P. Gille, M. Armbrüster, Yu. Grin, R. Schlögl, and O. Gröning, Langmuir. 28, 6848 (2012).