

ABSTRACT
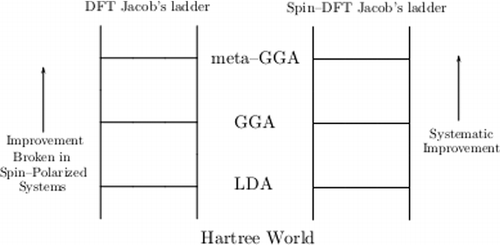
The ground-state density and energy of electrons in a spin-independent external potential are described exactly in principle by two related theories, density functional theory (DFT) and spin-density functional theory (SDFT), differing only in their use of the total density versus the up- and down-spin densities. With semilocal approximations to the exchange-correlation energy, the atomisation energies of molecules improve remarkably as we satisfy more exact constraints in SDFT, but not in DFT. We explain why only SDFT is amenable to semilocal approximation in spin-polarised systems such as typical open-shell atoms, and point out the historical importance of SDFT in chemistry. In spin-unpolarised systems, for which DFT and SDFT are equivalent, both theories are normally amenable to semilocal approximations which improve as more exact constraints are satisfied (predicting better equilibrium structures, energies, and energy differences).
GRAPHICAL ABSTRACT
Acknowledgements
John P. Perdew acknowledges discussions with Bradford B. Wayland. The authors are grateful for insightful discussions or collaborations with Andreas Savin.
Disclosure statement
No potential conflict of interest was reported by the authors.