
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Quantifying an atom's transferability, in a force field context, demands a quantitative understanding of how an atom ‘experiences’ the surrounding environment both intra-atomically and inter-atomically. Here we investigate the intra-atomic (EintraA) viewpoint through the study of the atoms Cα, Hα, N, O, and S in a series of ‘mono’-, tri- and penta-peptides. The remaining inter-atomic viewpoint consists of an electrostatic (via multipole moments), exchange and correlation components respectively, of which the electrostatic component has been previously reported. Together these four energy components, as calculated from the Interacting Quantum Atoms (IQA) partitioning approach, express the foundation of the Quantum Chemical Topological Force Field (QCTFF). In order to have transferability within a force field, smaller sample systems must be calculated and developed as representative of larger target systems.
The Cα, Hα, N, O and S atoms in a tri-peptide are energetically comparable to those in their penta-peptide configurations, within 2.1 kJ/mol in absolute value (1 exception). Across all five elements, this energy difference is on average ∼0.3 kJ/mol. On average, the tri-peptide sample systems represent a ∼8.2 Å atomic horizon around the central atoms of interest. Thus, both the previous knowledge of the ∼10.3 Å horizon sphere and ∼0.4 kJ/mol error required by the electrostatic multipole moments, determine how two of the four key QCTFF energy components are affected by an atom's molecular environment.
GRAPHICAL ABSTRACT
Dedication: It is a pleasure to contribute to a special issue in honour of Andreas Savin, a most generous host with whom I have enjoyed several discussions. As a deep and valiant thinker, a true scientist such as Andreas will surely continue to work in this capacity beyond his official retirement.
1. Introduction
Here we push computational boundaries to help answering an important but rather under-documented question. This question is of interest to anyone concerned [Citation1] with Quantum Chemical Topology (QCT) [Citation2–5] and focuses on the energetic transferability of a topological atom, now within the context of Interacting Quantum Atoms (IQA) [Citation6]. An answer to the question of energetic transferability is pivotal in the development of a force field because transferability is its zeroth cornerstone [Citation7]. All structure and dynamics predicted by a force field depend on the energy predictions it makes, and therefore we need to know the size of the atomic environment that still influences the energy of a central atom of interest.
A force field is asked to make predictions on a target system, which can be a protein in a large box of water, or a sizeable piece of RNA. Inevitably, target systems are large, and of course too large to serve as a sampling system for a force field. Even if this force field parameterisation were possible, it would defeat the object because it does not make sense to parameterise a dedicated force field for each target system. Instead, one parameterises a much smaller set, which is the sampling system. The question is then how small this sampling system can be. In the case of a protein as a target system, can the sampling system be a single amino acid? Or should it be a protein fragment consisting of three amino acids? Transferability studies can answer this question although perfect transferability is impossible: one cannot take a topological atom from a sampling system and literally transfer it to the target system without introducing an energetic error. So, really, transferability is not binary but a sliding scale, and we will discuss our data below keeping this in mind.
In the past, we have reported that ‘the true predictive power of a force field depends on the reliability of the information transfer of small molecules (or molecular clusters) to large molecules. Only if this transferability is high, a force field will make reliable predictions’ [Citation8]. The expression ‘information transfer’ is deliberately kept general and open in order to welcome various atomic properties as gauges of transferability. For example, in a study [Citation9] on the transferability of methylene and methyl fragments in alkylethers, atomic volumes (and even bond critical point properties) were invoked to quantify transferability. Because we believe that energy is the ultimate arbiter in force field transferability, we have studied properties more directly related to energy than volume, or intra-atomic energy itself, as we do in this paper. Given that interatomic electrostatic energy, at sufficiently long range, can be exactly represented by a multipole expansion [Citation10], it makes sense to study the transferability of atomic multipole moments. This was done in the context of computing atom types [Citation11], while the next study [Citation12] on transferability came one step closer to energy by studying the atomic electrostatic potential. Although useful, this type of assessment demands the construction of a grid at which the potential is evaluated. The necessity for such a grid is a vulnerability, which can be circumvented by going beyond the potential and investigating the interatomic electrostatic energy itself. However, this type of assessment introduces one or more atoms tasked to probe the central atom of interest. Such an investigation [Citation13] was done some time ago for a water trimer and a microhydrated serine. Amongst other findings, this study showed that the atoms of serine are more transferable, in going from the isolated serine to the Serine…(H2O)5 supermolecule, than the atoms in the water cluster.
Other research groups have also investigated the issue of transferability [Citation14–20]. The transferability question does not solely focus on the role of atomic multipole moments in the literature. The highly transferable nature of common structural properties such as bond lengths and angles has also been reported [Citation21].
2. Methodology
2.1. General background
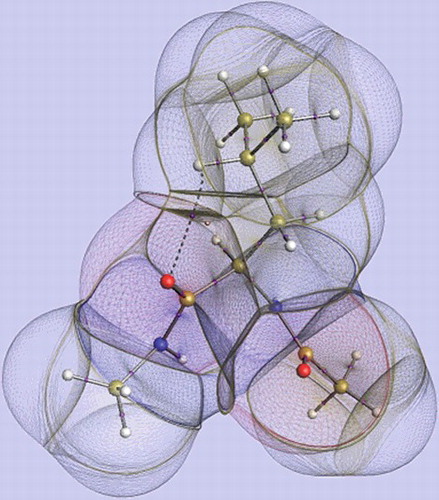
We strive towards the completion of a topological force field known as QCTFF [Citation22]. QCTFF will be a novel atomistic protein force field that builds on the principle of QCT, which is using the gradient vector field as a (minimal) means to let a quantum function partition itself in space. If this quantum function is the electron density then the product of the partitioning is a set of topological atoms. This was the first result of QCT, better known under the name Quantum Theory of Atoms in Molecules (QTAIM) [Citation3]. An example of topological atoms is shown in . Topological atoms are boxes of finite volume, with a peculiar shape that precisely reflects the whole quantum system they are part of. In other words, the whole system imprints its presence (at least in principle, not necessarily with much ‘numerical power’) onto each of the (topological) atoms within the system. We also note that there are no gaps [Citation23] between the atoms. This means that each little bit of electron density in a potentially large ‘pocket’ belongs to one atom or another. This means that the electrostatic potential this piece of electron density generates also belongs to an atom, always. As a further consequence, one can assert that all energy contributions (invariably associated with electron density) are always assigned to one atom or a pair of atoms. In short, all intra-atomic or interatomic energies are accounted for.
Figure 1. A representation of the topological atoms in Leucine, which is capped both at the N-terminus and the C-terminus by a peptide bond. The nuclear configuration is taken from a (Leu)5 conformer geometry-optimised at HF/6–31+G(d,p) level of theory.
QCTFF is built on the precise constellation of four resolutions [Citation22]. The first resolution is summarised by adopting the topological atom as the carrier of chemical information. Indeed, the total energy of a sampling system, and then ultimately a target system, can be predicted from the information that the atoms carry. The second resolution stipulates that the total energy is partitioned into four types of energy contributions, each of which has a chemical meaning: (1) intra-atomic self-energy, and interatomic (2) Coulomb, (3) exchange and (4) correlation energy. A short mathematical description of the IQA partitioning is provided in Section 2.3. This scheme is inspired by IQA, which in turn was inspired by the early establishment [Citation24] of a six-dimensional integration over two topological atoms simultaneously. This type of calculation returns the potential energy between two atoms, for any molecular geometry. This achievement makes the calculation of the intra-atomic energy independent of the atomic virial theorem [Citation25]. Third, any 1/r type of interaction can be expanded, thereby introducing spherical harmonic (atomic) multipole moments. This expansion is of great practical use for the Coulomb energy, provided that the multipole expansion converges [Citation26,Citation27]. Finally, a variation in nuclear configuration causes a change in a given atom's energies and its multipole moments. The machine learning method Kriging [Citation28,Citation29] has the capacity to capture the mapping between the coordinates of the atom's surrounding nuclei (input) and this atom's energies and multipole moments (output). The input is cast into a number of so-called features, the details of which are described elsewhere [Citation29]. It suffices to state here that features are essentially internal geometrical coordinates that allow a given atom to describe its own atomic environment (that is, basically by means of nuclear positions). Note that a Kriging model is trained on a data set of sample systems.
When each of the four aforementioned energies undergoes Kriging, then a complete molecular energy Kriging model is generated. Compiling these models, along with other comparable models from other molecules into a database, results in the ultimate formation of QCTFF. At short range, the electrostatic energy (or Coulomb energy without being pedantic) between two atoms can only be calculated directly, that is, without using multipole moments. These interatomic energies then serve as output for a Kriging model. However, at long-range, a convergent multipole expansion is used and each Kriging model takes up the multipole moment as its output. So far, most work has been done [Citation30-35] on multipole moments, up to hexadecapole in fact.
The exchange energy can also be expanded into so-called exchange (multipole) moments as first demonstrated [Citation36] in 2007. However, these moments carry imprints of the molecular orbitals themselves, a difficulty that has not been overcome. This is why we instead follow the route of unexpanded exchange energies. Fortunately, for saturated systems [Citation37] (and many non-metallic condensed matter systems are saturated) these energies drop off very quickly with distance. The local nature of the exchange energies makes it feasible to krige them as energies. Locality means that a given atom does not have to be aware of too deep an environment. Because only the immediate neighbours suffice, the number of possible nuclear configurations around the given atom is restricted. Indeed, atoms that are covalently attached to a given atom cannot move around as much compared with atoms further away (that are hence less covalently attached).
Dynamical correlation energies were first [Citation38] calculated through coupled cluster theory, and can also be offered to a Kriging engine to then be mapped onto the coordinates of the surrounding atoms. Work is in progress to do this for inter-atomic correlation energies from Møller–Plesset wave functions. However, the first non-electrostatic energy that was ever kriged is the atomic kinetic energy [Citation39]. Building on this success more non-electrostatic energy components have been kriged in our lab and will be published in due course. However, the subject of this paper is not Kriging but the transferability of a non-electrostatic energy, namely the intra-atomic energy or self-energy, for short. This paper will report the results seen for the intra-atomic self-energy in a series of oligopeptides of varying length.
The intra-atomic energy represents the energy that an atom possesses inside a molecular system. Understanding the energetic cost of transferring an atom from a smaller sampling system into a larger target system reveals whether the sampling system is suitable to be used as a sample for this target. If the sampling system is too small then one observes a large change in self-energy between the given atom in the sampling system as compared with that atom in the target system. An assessment of the atom in the sample system and then in the target system results in a quantitative measure of their similarity. This similarity measure enables the computation of atom types, an achievement that was realised some time ago [Citation11] but based on atomic properties such as multipole moments, virial-based atomic energy and volume. In future work, the intra-atomic energy of an atom could add value and play a role in the classification and computation of atom types, which is under-researched.
2.2. Horizon sphere, atomic horizon and compounds studied
The case studies chosen for the analysis focus on homogeneous oligopeptides of three possible sizes: mono- (n = 1), tri- (n = 3) and penta-peptide (n = 5), where n is the number of amino acids in the peptide chain. Eight systems are analysed in total, seven of which (Alanine, Glycine, Isoleucine, Leucine, Serine, Threonine and Valine) will be used to investigate the transferability of the central α-carbon, α-hydrogen, amino-oxygen and amino-nitrogen atoms. The eighth system (cysteine) is used to investigate the transferability of a sulphur sidechain atom. This investigation is analogous to that in a previous paper featuring the study of the same five elements (C, H, O, N and S) in the small and naturally occurring protein crambin [Citation40]. That study introduced the concept of a horizon sphere. This concept addresses the following simple question: For a selected central atom, how do its neighbours influence its multipole moments and at which distance can their influence be ignored?
This horizon sphere was presented as a metaphorical sphere, which when centred on a single atom, correlates the energetic change in an atom's multipole moment with the sphere's radius. This allows the intuitive mapping of two commonly known physical parameters (interaction energy and distance). Operationally, the horizon sphere incrementally increases its radius in steps of 0.1 Å and observes the growing number of other atoms appearing within its volume. At every step, new atoms may enter the sphere. If not, the horizon sphere grows by another step. As such, a set of nested atomic configurations appears, each containing the central atom, and one can observe how the multipole moments of the central atom change with increasing configuration size. In other words, the horizon sphere allows a chemically meaningful measurement of how far out the central atom still experiences the presence of its atomic environment. In short, how far does the given atom ‘feel’?
In crambin, the largest structure considered had a radius of 12 Å, and was taken as the reference structure. It consisted of 294 atoms in total, while four atoms (one of each element or C, H, O and N) were selected as ‘probing atoms’. The multipole moments of the latter always remained invariant. These multipole moments were combined with the varying multipole moments of the central atom in the horizon sphere to yield the electrostatic interaction energy between that atom and a probing atom.
The conclusion from the crambin work was that each element had its own horizon sphere radius (Cα = 10.5 Å, Hα = 7.7 Å, O = 11.0 Å, N = 11.3 Å and S = 10.8 Å) where each atom's multipole moments are influenced by the presence of other atoms (by no more than 0.4 kJ/mol). The current study expands and complements the crambin study from the perspective of atomic self-energy. The latter produces its own horizon sphere radii, as will become clear later. In the presence of two types of radii, the question is then how to quantify transferability. One way would be to take the larger radius of the two because the larger radius is the most ‘demanding’ in terms of transferability. However, one could also argue that ultimately a force field adds the various energy contributions that it uses to describe a system, and hence the sum of the energies needs to be screened for transferability. In this paper, we will not follow either option but focus on the transferability of the self-energy itself. We know from unpublished work (on different systems, i.e. large water clusters) that the exchange energy generates smaller horizon radii. This is not surprising in the light of previously published work [Citation37], which shows how quickly exchange energy drops off with distance (for saturated systems).
In retrospect, the horizon sphere would have been better called atomic horizon thereby allowing it to be not spherical, in general. In this work we do not construct a horizon sphere but control the size of the sample system by varying the length of an oligopeptide. This size control occurs ‘linearly’ in that the chain length of the oligopeptide is varied, which is reminiscent of the primary structure of a protein. However, the oligopeptide may very well curl up, reminding us of the importance of secondary structure. The largest size oligopeptide then represents a globular environment with respect to which a given atom is studied. The edge of this environment is more accurately called an atomic horizon. One can then introduce a ‘pseudo’-radius for this atomic horizon by measuring the distance between the nuclear positions of the atom of interest and the atom furthest away from it.
2.3. Interacting quantum atoms (IQA): some relevant formulae
The IQA partitioning quantitatively describes the energetic of topological atomic, that is, their interaction as well as their internal energy, through a combination of kinetic and potential energies [Citation6]. These energies consist of an intra-atomic or an interatomic contribution. At an unrefined level, the IQA formalism partitions the molecule's energy according to Equationthe(1)
(1) following equation:
(1)
(1)
The energy term EintraA can be broken down in three contributions:
(2)
(2) where TA is the kinetic energy of the electrons, VeeAA is the electron–electron repulsive potential energy and VenAA is the attractive electron-nuclear potential energy, all within atom A. Note that the kinetic energy is well defined for the topological atom, which would not be true for an arbitrary (atomic) subspace. Together, these three energy contributions comprise the self-energy possessed by a single atom. This energy is the central quantity investigated in the present work. For completeness, the remaining interatomic energy attributed to an atom is defined in the following equation:
(3)
(3) where VenAB, VneAB and VeeAB are as described above but with respect to both A and B. The quantity VnnAB is the repulsive nuclear–nuclear potential energy, which is totally classical within the Born–Oppenheimer approximation. For the sake of completeness, the VeeAB contribution can be specified further,
(4)
(4) where the first term on the right-hand side embodies the Coulomb interaction between electrons in atoms A and B, the second term is the electronic exchange energy (between A and B) while the third term is the most challenging term to calculate, which is associated with dynamic correlation or dispersion.
A further rearrangement of the energies take places in EquationEquation (5)(5)
(5) , and following this, a new expression for the complete interaction energy between two atoms, denoted VinterAB, can then be formed, as
(5)
(5)
(6)
(6)
Here, VelecAB represents the complete electrostatic interaction energy between two atoms A and B, now including the interaction with the respective nuclei. This quantity (rather than VABCoul) is the energy that has been expanded as a multipolar series on many occasions [Citation26,Citation27,Citation40–44] in the past.
For a more exhaustive description of the partitioning scheme including additional formulae and previous applications, the reader is directed to the original literature by Blanco et al. [Citation6,Citation38,Citation45–48]. For the purpose of this paper, we will only present the transferability assessment from the point-of-view of the intra-atomic energy EintraA as defined in EquationEquation (2)(2)
(2) .
2.4. Computational details
The penta-peptide geometries were the result of a geometry optimisation at the HF/6-31+G(d,p) theory level using the GAUSSIAN09 program [Citation49]. No frequency calculations were carried out because confirming that the optimised geometries are true energy minima is not essential in reaching the conclusion of this paper, and the cost of these extra calculations is huge, given their size. A randomly generated penta-glycine (Penta-gly) was created from scratch by the program Gaussview and its sidechain changed according to the amino acid to be analysed. For each of the eight oligopeptides studied, the penta-peptide always provided the exact geometry of the tri-peptide and the single amino acid, that is, for the atoms the penta-peptide (n = 5) has in common with its derivatives (n = 3 or n = 1). Hence, the tri-peptide and the single amino acid were not geometry-optimised. Thus, the current transferability study freezes out any geometry changes while comparing the penta-peptide with the tri-peptide, for example. Note that neither di-peptides nor tetra-peptides were included in this study (i.e. n = 2 or n = 4). They are excluded to ensure that the radius decreases approximately symmetrically at either side of the central amino acid under study.
Following the optimisation of the penta-peptide, this system is first trimmed to form the corresponding tri-peptide and then again to form the single amino acid. When trimming the N-terminus, the CH3NHC( = O)– group is removed and replaced by a hydrogen atom. This means that the first α-carbon of the original penta-peptide now becomes a methyl group. This methyl group caps the emerging tri-peptide in the same way as the now removed methyl capped the original penta-peptide. Similarly, trimming at the C-terminus means removing the CH3C(= O)NH– group and replacing it by a hydrogen. Again, this substitution generates a terminal methyl group, which is part of the newly formed but familiar CH3NHC(= O)– capping group. The geometrical positions of hydrogen atoms in these methyl caps are predetermined by previous atomic positions in the larger chain. Their bond lengths are standardised to be 1.07 Å in accordance with the standard parameters in Gaussview.
The IQA energy partitioning was carried out by the program AIMAll [Citation50–52]. Default inputs were used. The keyword ‘encomp = 3’ was included in the input, which is the short-hand input name referring to ‘Energy Components’. Some poorly calculated atoms were recomputed with a specified outer angular quadrature (‘skyhigh_lebedev’) for the atom under study, instead of the default ‘auto’ in an attempt to obtain a more accurate calculation. This improved some atomic integration errors, but also worsened some. The best from both sets of runs were selected for further analysis. Typical CPU times for oxygen atoms (which took most time compared to other elements) in penta-peptides amounted to up to 24 hours on 32 cores, highlighting the compute intense nature of the current study. Table S1 in the Supplementary Information gives an impression of the general atomic integration accuracy obtained from the L(Ω) value [Citation53], which settles for about 0.2–0.5 kJ/mol for all elements except carbon where L(C) = 2.0 kJ/mol. The latter error is worrisome but could not be improved in spite of several attempts.
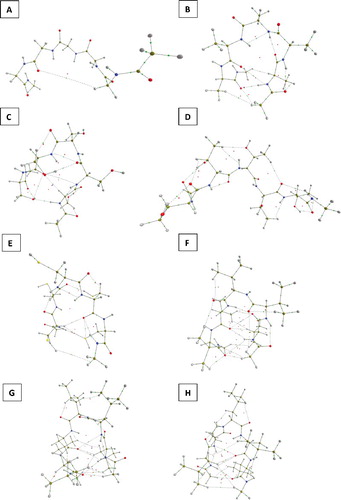
With regard to visualisation, was generated with in-house software called IRIS, which is based on earlier work [Citation54,Citation55]. The nuclear configuration is taken from a (Leu)5 conformer geometry-optimised at HF/6–31+G(d,p) level of theory (51 Molecular Orbitals and 542 Gaussian primitives). This figure represents mono-Leucine whose geometry was taken directly from the optimised penta-Leucine (the central amino group ‘3’) and capped by the familiar CH3NH– group at the C-terminus and CH3C( = O)– group at the N-terminus. The wave function was again calculated at HF/6–31+G(d,p) level. IRIS's default settings were employed, other than using wireframe for the surface and altering the transparency. Default element colours were used. The images of each of the eight penta-peptides (geometry-optimised at HF/6–31+G(d,p) level) shown in were created by AIMStudio [Citation50]. Each penta-peptide is capped at both termini by the same groups as in . Default settings were used other than changing the electron density cutoff to 1×10−6 a.u. in order to ensure that there are no ‘gaps’ in the non-covalent interaction lines. Each molecule was viewed individually and screenshot. The eight screenshots were combined to give the final image in
Figure 2. (A) Penta-Gly, (B) Penta-Ala, (C) Penta-Ser, (D) Penta-Thr, (E) Penta-Cys, (F) Penta-Val, (G) Penta-Leu and (H) Penta-Ile configurations (suspected as local energy minima but not confirmed through frequency calculations).
3. Results and discussion
Here we monitor how the EintraA energies of each of the five elements occurring in naturally amino acids change with peptide size. The hypothesis for the overall analysis is that the central atoms of the penta-peptides will be closest in energy to the corresponding central atoms in the tri-peptides. If the hypothesis is true, within a suitable energy margin, then the tri-peptide atoms can sufficiently accurately represent (or model) the atoms in the (penta-peptide) target system.
shows the precise configurations of each of the eight penta-peptides investigated. The geometry of these peptide chains are not constrained or biased to any conformations during the ab initio geometry optimisation and are all initialised in a consistent and random way. Hence, no optimisation is directed towards a predominant favourable growth pattern, e.g. ‘linear’ in one direction. Linear oligopeptides are like open chains, i.e. extended in one dimension. On average, the internuclear distances in such open configurations are larger, as opposed to ‘curled’ configuration where the central atom is surrounded by atoms that are closer by. One expects more distant atoms (in the environment) to have less influence on the central atom. Therefore, the central atom will be more readily transferable, since most of its environment in the penta-peptide does not matter much. Hence, a transferability test on an open (i.e. linear, extended) configuration is less severe than one on a curly (i.e. globular) configuration. The majority of the configurations are curly, so our transferability tests are severe. Finally we note that, although not directly investigated here, it is well known that the number of local energy minima present in oligopeptide conformational space is vast [Citation56]. Based on the data presented here, we cannot be sure that the fixed configurations studied are representative for configurations found in the Protein Data Bank, for example. However, the uniform treatment and the lack of bias gives some comfort that the results may be universal.
Across the eight geometry-optimised penta-peptides, the penta-Gly system showed a preference for helical growth (in the peptide chain). However, the penta-Thr system showed no obvious preference in adopting a pronounced secondary structure, while the remaining six penta-peptides curl up to form conformations resembling β-turns. In all penta-peptides, numerous intra-molecular interactions were observed, marked by a complex network of bond and ring critical points. These intramolecular interactions also increase the possibility for a network of interactions that connects the atoms of the central amino acid to the termini of the oligopeptide. This phenomenon is prevalent in water clusters and known to influence atomic energies [Citation57,Citation58]. This effect adds another dimension of complexity to the study.
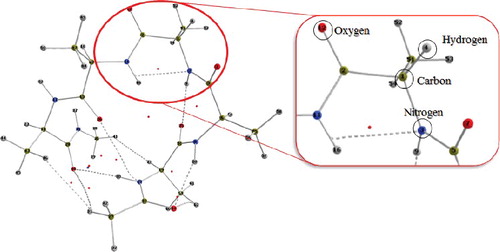
clarifies the actual atoms studied, each representing an element. Clearly the atoms appear in the third amino acid, in the middle of the penta-peptide, or in the middle of the tri-peptide, or as the atoms of the single amino acid of course.
Figure 3. Penta-Ala system. Highlighted atoms represent the central atoms under study.
The differences in self-energies (EintraA see EquationEquation (2)(2)
(2) ) for each element across each peptide system are summarised in (for nitrogen), (for oxygen), (for carbon), (for hydrogen) and (for sulphur). Each table details the difference in intra-atomic energies, denoted ΔE, for the given atom across the sequence of three oligopeptides (‘penta’, ‘tri’ and ‘mono’), one entry for each of the eight amino acids (except cysteine). The data of the cysteine oligopeptides are only used to study the element sulphur. The energy differences listed in all the tables are very much smaller than the typical magnitude of the EintraA energies for the five possible elements, which are huge: nitrogen ∼−140,000 kJ/mol, oxygen ∼−195,000 kJ/mol, carbon ∼−100,000 kJ/mol, hydrogen ∼−1200 kJ/mol and sulphur ∼−1,042,000 kJ/mol, respectively.
Table 1. Energy differences (ΔE) in peptidic nitrogen (see ) intra-atomic energies. All energies are in kJ/mol.
Table 2. Energy differences (ΔE) in carbonyl-oxygen (see ) intra-atomic energies. All energies are in kJ/mol.
Table 3. Energy differences (ΔE) in α-carbon (see ) intra-atomic energies. All energies are in kJ/mol.
Table 4. Energy differences (ΔE) in α-hydrogen (see ) intra-atomic energies. All energies are in kJ/mol.
Table 5. Energy differences (ΔE) in side-chain sulphur intra-atomic energies. All energies are in kJ/mol.
First, the nitrogen and oxygen results confirm the hypothesis stated above, that the energy difference between the tri-and penta-peptide (i.e. ΔE2) is the smallest possible of the three energy differences. The maximum energy difference (in absolute value) between the penta-peptide and tri-peptide self-energy for N is 3.3 kJ/mol, and 0.8 kJ/mol for O. Overall, combining all entries of oxygen and nitrogen, in 11 of the 14( = 2×7) cases, energy differences are smaller than 1 kJ/mol. This is a very pleasing result, considering the magnitude of the intra-atomic energies of these atoms. However, this result also highlights the accuracy required for a study of this nature. The average integration errors (L(Ω), given in the fifth columns of each table), validate this conclusion. The reported errors are an average of the integration error calculated for the atom in each of the three chain lengths. Overall, the total intra-atomic energy differences across the olidopeptides are below 0.0016% and 0.0012%, respectively.
Observing the trends for the α-carbon and its bonded α-hydrogen atom, the message is less pleasing. For the carbon atoms the hypothesis only holds for 4 (Ala, Thr, Val and Ile) of the 7 amino acids. The same is true for the α-hydrogens (Ala, Thr, Gly and Leu). However, for both Cα and Hα, much smaller total energy differences are observed across all oligopeptides, in general. For carbon and hydrogen, absolute energy differences fall under 3.9 kJ/mol (two exceptions: Ala and Ile) and 0.65 kJ/mol (one exception: Ala), respectively. These small energy differences combined with the relatively much larger averaged atomic integration errors L(Ω), make for less convincing results. It appears that, when the averaged integration error has a magnitude similar to that of the energy difference, then the hypothesis does not hold. When the average integration error is sufficiently smaller than the energy difference, then the hypothesis holds. Perhaps fortuitously, some hydrogen energy differences still confirm the hypothesis, despite the integration errors being relatively large in comparison to each of the energy differences (e.g. Thr ΔE2 = 0.14 (±0.4) kJ/mol and Gly ΔE2 = −0.01 (±0.3) kJ/mol)). However, there are still three cases (Ser, Val and Ile) where the integration error is the accuracy limiting factor. The integration errors observed throughout the hydrogen atom analysis are smaller than 0.4 kJ/mol in absolute value, and would normally be considered very accurate. However, for these atoms, the difference in the EintraA energy is very low and in most cases is smaller than the mean error. The same observations can be made for the α-carbons, which are known to be more difficult to accurately integrate due to the increased complexity due to its tetrahedral hybridisation [Citation59]. The hybridisation of the atom generally appears to correlate well with the accuracy of the calculation (see Table S1). The atomic integration errors are not a problem for the oxygen atoms and only a minor effect for the nitrogen atoms. Overall, these atoms have the optimal balance of good atomic integration errors and large EintraA energetic differences (>5 kJ/mol in absolute value).
Finally, shows that the sulphur atom in the Cys oligopeptide chains also conforms to the expected trend with an energy difference, all in absolute value terms of 1.67 kJ/mol between tri- and penta-chains (ΔE2) as compared with 7.10 kJ/mol and 8.78 kJ/mol for ΔE1 and ΔE3, respectively.
Tables S2–S6 of the Supplementary Information summarise the pseudo-radii of the atomic horizons of each of the elements in each of the oligiopeptides studied. Previous work on crambin [Citation40] showed that the electrostatic multipole moments generate a unique horizon sphere radius for each element. lists these radii alongside the atomic horizon pseudo-radii obtained here for the averaged EintraA values of the tri-peptides.
Table 6. Horizon sphere radii (for multipolar electrostatics) and atomic horizon pseudo-radii (for EintraA) in each element in crambin and the oligopeptides. ΔE values are also provided for these radii in kJ/mol. Data for the multipole moments were taken from Ref. [Citation40] and data for the oligopeptides are averaged across all tri-peptides.
It is clear that, for each element, the EintraA causes a smaller atomic horizon compared to the horizon sphere of the multipole moments. The one exception to this conclusion is Hα, for which the multipolar horizon sphere radius is smaller, by 0.6 Å. However, as we have observed the Hα atoms to show very little energetic change across all mono-, tri- and penta-peptide chain lengths (|ΔE | approx. < 0.3 kJ/mol), we believe it is fair to treat the ‘mono-peptide’ (i.e. single amino acid) as a suitable sample system size for these atoms. Hence, a new atomic horizon pseudo-radius can be calculated for Hα only, based on the mono-peptide spheres. The mono-peptide averaged atomic horizon pseudo-radius is 5.2 Å for the Hα atoms, which coincides with observing the smaller EintraA pseudo-radius compared to the multipolar horizon sphere radius.
The energy differences associated with EintraA are smaller than those associated with the multipole moments. In general, an energy difference (whether from multipole moments or EintraA) becomes smaller and smaller with increasing size of the sample system. If this energy difference reaches zero, one can conclude that convergence occurs. Comparing ΔE1 (TRI-MONO) and ΔE2 (PENTA-TRI) gives an impression of the speed of convergence. Indeed, if ΔE2 (PENTA-TRI) << ΔE1 (TRI-MONO) then the convergence is fast. From the respective tables it is clear the N, O and S atoms show fast convergence. However, the Cα atom and Hα atom converges slower. We note that their ΔE1 (TRI-MONO) values were already quite small in the first place. Returning to sulphur, it is regarded as fast converging despite a large ΔE2 (PENTA-TRI). This fact can be rationalised by the larger number of electrons that a sulphur atom owns and, thus, the greater the complexity and sensitivity of the EintraA energy. In general, the convergence is faster for EintraA compared the multipolar energies (studied in crambin [Citation40]), as can be seen from . In addition, the atomic horizons of the various elements are also smaller for the EintraA as compared with those of the multipolar energies.
4. Conclusion
The atomic self-energy, denoted EintraA, is studied as a gauge of energetic transferability for five elements (H, C, N, O and S) occurring in homogeneous oligopeptides (of increasing length) of eight possible amino acids. The self-energy of a given atom is systematically monitored as a function of a chemical environment growing in size but while freezing the geometry of the central amino acid.
The central hypothesis of the current work is that EintraA of an atom in a tri-peptide is quantitatively close to EintraA of the corresponding atom in the penta-peptide. This hypothesis proves unreservedly correct for the oxygen, nitrogen and sulphur atoms. However, for the α-carbon atoms the hypothesis is harder to prove because the atomic integration errors are larger than for the other four elements. The α-hydrogen atoms show very small differences in EintraA across all three sizes of (oligo)peptide. However, for α-hydrogen and α-carbon overall, the central hypothesis is true in 8 out of 14 (= 2×7) peptides. For the remaining six cases, the atomic integration errors are too large to be conclusive.
We have learned that for the elements studied, the tri-peptide is a sufficient sample system to accurately predict the energy (on average to within ∼0.32 kJ/mol) for the corresponding element in the target penta-peptide. For hydrogen, the sample size can be reduced further to a single amino acid as a result of small energy differences (|ΔE | approx. < 0.3 kJ/mol) across all oligopeptides.
The convergence of the intra-atomic energy is generally faster compared to multipole moments. In addition, the atomic horizon pseudo-radii are smaller than the radii of the multipolar electrostatics horizon spheres.
Atomic integration errors will ever improve with algorithmic efficiency and, thus too, will the accuracy of the partitioned energies of a molecule. Future work will involve the study of the EIQAA component for a complete description of the atomic horizon. This will provide supplementary resource and knowledge towards the development of QCTFF.
Supplemental data
Supplemental data for this article can be accessed at http://dx.doi.org/10.1080/00268976.2015.1116717.
tmph_a_1116717_sm0981.docx
Download MS Word (27.6 KB)Acknowledgements
P. Maxwell thanks the BBSRC for funding his PhD studentship and P.L.A. Popelier thanks the EPSRC for funding his Established Career Fellowship.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- P.L. Ayers, R.J. Boyd, P. Bultinck, M. Caffarel; R. Carbó-Dorca, M. Causá, J. Cioslowski, J. Contreras-Garcia, D.L. Cooper, P. Coppens, C. Gatti, S. Grabowsky, P. Lazzeretti, P. Macchi, A.M. Pendás, P.L.A. Popelier, K. Ruedenberg, H. Rzepa, A. Savin, A. Sax, W.E.H. Schwarz, S. Shahbazian, S. Silvi, M. Solà and V. Tsirelson, Six questions on topology in theoretical chemistry. Comput. Theor. Chem. 1053, 2 (2015).
- R.F.W. Bader, S.G. Anderson and A.J. Duke, Quantum Topology of Molecular Charge Distributions. J. Am. Chem. Soc. 101, 1389(1979).
- R.F.W. Bader, Atoms in Molecules: A Quantum Theory (Oxford Univ. Press, Oxford, Great Britain, 1990).
- P.L.A. Popelier and É.A.G. Brémond, Geometrically faithful homeomorphisms between the electron density and the bare nuclear potential. Int. J. Quant. Chem. 109, 2542 (2009).
- P.L.A. Popelier, The Quantum Theory of Atoms in Molecules, in The Nature of the Chemical Bond Revisited, edited by G. Frenking and S., Shaik (Wiley-VCH, 2014), Chapter 8, pp. 271–308.
- M.A. Blanco, A.M. Pendas and E. Francisco, Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theor. Comput. 1, 1096 (2005).
- P.L.A. Popelier, A generic force field based on quantum chemical topology, in Modern Charge-Density Analysis, edited by C. Gatti and P. Macchi (Springer, Germany, 2012), Vol. 14, pp. 505.
- P.L.A. Popelier, Quantum Chemical Topology: Knowledgeable Atoms in Peptides. AIP Conf. Proc. 1456, 261 (2012).
- A. Vila and R.A. Mosquera, Transferability in alkyl monoethers. II Methyl and methylene fragments. J. Chem. Phys. 115, 1264 (2001).
- S. Cardamone, T.J. Hughes and P.L.A. Popelier, Multipolar Electrostatics. Phys. Chem. Chem. Phys. 16, 10367 (2014).
- P.L.A. Popelier and F.M. Aicken, Atomic Properties of Amino Acids: computed Atom Types as a Guide for future Force Field Design. ChemPhysChem 4, 824 (2003).
- P.L.A. Popelier, M. Devereux and M. Rafat, The quantum topological electrostatic potential as a probe for functional group transferability. Acta Cryst. A60, 427 (2004).
- M. Rafat, M. Shaik and P.L.A. Popelier, Transferability of quantum topological atoms in terms of electrostatic interaction energy. J. Phys. Chem. A. 110, 13578 (2006).
- C.H. Faerman and S.L.A. Price, Transferable Distributed Multipole Model For the Electrostatic Interactions of Peptides and Amides. J. Am. Chem. Soc. 112, 4915 (1990).
- W.T.M. Mooij, F.B. van Duijneveldt, J.G.C.M. van Duijneveldt-vande Rijdt and B.P. van Eijck, Transferable ab Initio Intermolecular Potentials. 1. Derivation from Methanol Dimer and Trimer Calculations. J. Phys. Chem. A. 103, 9872 (1999).
- K.E. Laidig, General Expression For the Spatial Partitioning of the Moments and Multipole Moments of Molecular Charge-Distributions. J. Phys. Chem. 97, 12760 (1993).
- C.E. Whitehead, C.M. Breneman, N. Sukumar and M.D. Ryan, Transferable atom equivalent multicentered multipole expansion method. J. Comp. Chem. 24, 512 (2003).
- C.M. Breneman, T.R. Thompson, M. Rhem and M. Dung, Electron density modeling of large systems using the transferable atom equivalent method. Comput. Chem. 19, 161 (1995).
- M. Woińska and P.M. Dominiak, Transferability of Atomic Multipoles in Amino Acids and Peptides for Various Density Partitions. J. Phys. Chem. A 117, 1535 (2011).
- S. Grabowsky, R. Kalinowski, M. Weber, D. Foerster, P. Carsten and P. Luger, Transferability and reproducibility in electron density studies – bond-topological and atomic properties of tripeptides of the type L-alanyl-X-Lalanine. Acta Cryst. B65, 488 (2009).
- Y. Yuan, M.J.L. Mills, P.L.A. Popelier and F. Jensen, Comprehensive Analysis of Energy Minima of the 20 Natural Amino Acids. J. Phys. Chem. A. 118, 7876 (2014).
- P.L.A. Popelier, QCTFF: On the Construction of a Novel Protein Force Field. Int. J. Quant. Chem. 115, 1005 (2015).
- P.L.A. Popelier, New insights in atom-atom interactions for future drug design. Current Topics in Med. Chem. 12, 1924 (2012).
- P.L.A. Popelier and D.S. Kosov, Atom-atom partitioning of intramolecular and intermolecular Coulomb energy. J. Chem. Phys. 114, 6539 (2001).
- R.F.W. Bader, Beddall P.M. Virial, Field Relationship for Molecular Charge Distributions and the Spatial Partitioning of Molecular Properties. J. Chem. Phys. 56, 3320 (1972).
- M. Rafat and P.L.A. Popelier, Long range behaviour of high-rank topological multipole moments. J. Comput. Chem. 28, 832 (2007).
- M. Rafat and P.L.A. Popelier, A convergent multipole expansion for 1,3 and 1,4 Coulomb interactions. J. Chem. Phys. 124, 144102-1-7 (2006).
- C.E. Rasmussen and C.K.I. Williams, Gaussian Processes for Machine Learning (The MIT Press, Cambridge, MA, 2006).
- G. Matheron, Principles of Geostatistics. Econ. Geol 58, 21 (1963).
- M.J.L. Mills and P.L.A. Popelier, Intramolecular polarisable multipolar electrostatics from the machine learning method Kriging. Comput. Theor. Chem. 975, 42 (2011).
- M.J.L. Mills and P.L.A. Popelier, Polarisable multipolar electrostatics from the machine learning method Kriging: an application to alanine. Theor. Chem. Acc. 131, 1137 (2012).
- C.M. Handley, G.I. Hawe, D.B. Kell and P.L.A. Popelier, Optimal Construction of a Fast and Accurate Polarisable Water Potential based on Multipole Moments trained by Machine Learning. Phys. Chem. Chem. Phys. 11, 6365 (2009).
- Y. Yuan, M. Mills and P.L.A. Popelier, Multipolar electrostatics based on the Kriging machine learning method: an application to serine. J. Mol. Model 20, 20 (2014).
- T. Fletcher, S.J. Davie and P.L.A. Popelier, Prediction of Intramolecular Polarization of Aromatic Amino Acids Using Kriging Machine Learning. J. Chem. Theory Comput. 10, 3708 (2014).
- S.M. Kandathil, T.L. Fletcher, Y. Yuan, J. Knowles and P.L.A. Popelier, Accuracy and Tractability of a Kriging Model of Intramolecular Polarizable Multipolar Electrostatics and Its Application to Histidine. J. Comput. Chem. 34, 1850 (2013).
- M. Rafat and P.L.A. Popelier, Topological atom-atom partitioning of molecular exchange energy and its multipolar convergence, in Quantum Theory of Atoms in Molecules, edited by C.F. Matta and R.J. Boyd (Wiley-VCH, Weinheim, Germany, 2007), Vol. 5, pp. 121–140.
- M. Garcia-Revilla, E. Francisco, P.L.A. Popelier and A.M. Martin-Pendas, Domain-averaged exchange correlation energies as a physical underpinning for chemical graphs. ChemPhysChem. 14, 1211 (2013).
- R. Chávez-Calvillo, M. García-Revilla, E. Francisco, A. Martín Pendás and T. Rocha-Rinza, Dynamical correlation within the Interacting Quantum Atoms method through coupled cluster theory. Comput. Theor. Chem. 1053, 90 (2015).
- T.L. Fletcher, S.M. Kandathil and P.L.A. Popelier, The prediction of atomic kinetic energies from coordinates of surrounding atoms using kriging machine learning. Theor. Chem. Acc. 133, 1499:1 (2014).
- Y. Yuan, M.J.L. Mills and P.L.A. Popelier, Multipolar Electrostatics for Proteins: Atom-Atom Electrostatic Energies in Crambin. J. Comp. Chem. 35, 343 (2014).
- M. Rafat and P.L.A. Popelier, The electrostatic potential generated by topological atoms. Part II: inverse multipole moments. J. Chem. Phys. 123, 204103-1,7 (2005).
- M. Rafat and P.L.A. Popelier, Atom-atom partitioning of total (super)molecular energy: the hidden terms of classical force fields. J. Comput. Chem. 28, 292 (2007).
- L. Joubert and P.L.A. Popelier, The prediction of energies and geometries of hydrogen bonded DNA base-pairs via a topological electrostatic potential. Phys. Chem. Chem. Phys. 4, 4353 (2002).
- L. Joubert and P.L.A. Popelier, Improved Convergence of the ‘Atoms in Molecules’ Multipole Expansion of Electrostatic Interaction. Mol. Phys. 100, 3357 (2002).
- K. Eskandari and C. Van Alsenoy, Hydrogen–Hydrogen Interaction in Planar Biphenyl: A Theoretical Study Based on the Interacting Quantum Atoms and Hirshfeld Atomic Energy Partitioning Methods. J. Comput. Chem. 35, 1883 (2014).
- J. Dillen, Congested Molecules. Where is the Steric Repulsion? An Analysis of the Electron Density by the Method of Interacting Quantum Atoms. Int. J. Quant. Chem. 113, 2143 (2013).
- A.M. Pendás, M.A. Blanco and E. Francisco, The nature of the hydrogen bond: A synthesis from the interacting quantum atoms picture. J. Chem. Phys. 125, 184112-1-20 (2006).
- A.M. Pendas, E. Francisco and M.A. Blanco, Binding Energies of First Row Diatomics in the Light of the Interacting Quantum Atoms Approach. J. Phys. Chem. A 110, 12864 (2006).
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.J.A. Montgomery, J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N.J. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, Ö. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski and D.J. Fox, Gaussian 09 (Gaussian, Inc., Wallingford, CT, 2009).
- T.A. Keith, AIMAll (TK Gristmill Software, Overland Park, KS, 2014; aim.tkgristmill.com).
- T.A. Keith, AIMAll (Version 13.10.19) (TK Gristmill Software, Overland Park, KS, 2013; http://aim.tkgristmill.com).
- T.A. Keith, AIMAll. 14.11.23 (TK Gristmill Software, Overland Park, KS, 2014; http://aim.tkgristmill.com).
- F.W. Biegler-Koenig, R.F.W. Bader and T.H. Tang, Calculation of the Average Properties of Atoms in Molecules. J. Comp. Chem. 1982, 3, 317.
- M. Rafat and P.L.A. Popelier, Visualisation and integration of quantum topological atoms by spatial discretisation into finite elements. J. Comput. Chem. 28, 2602 (2007).
- M. Rafat, M. Devereux and P.L.A. Popelier, Rendering of quantum topological atoms and bonds. J. Mol. Graphics Modell. 24, 111 (2005).
- W. Yu, Z. Wu, H. Chen, X. Liu, A.D. MacKerell J. and Z. Lin, Comprehensive Conformational Studies of Five Tripeptides and a Deduced Method for Efficient Determinations of Peptide Structures. J. Phys. Chem. B 116, 2269 (2012).
- H. Lu, Y. Wang, Y. Wu, P. Yang, L. Li and S. Li, Hydrogen-bond network and local structure of liquid water: An atoms-in-molecules perspective. J. Chem Phys 129, 124512 (2008).
- J.M. Guevara-Vela, R. Chavez-Calvillo, M. Garcia-Revilla, J. Hernandez-Trujillo, O. Christiansen, E. Francisco, A.M. Pendas and T. Rocha-Rinza, Hydrogen-Bond Cooperative Effects in Small Cyclic Water Clusters as Revealed by the Interacting Quantum Atoms Approach. Chem. Eur. J. 19, 14304 (2013).
- F.M. Aicken and P.L.A. Popelier, Atomic properties of selected biomolecules. Part 1. The interpretation of atomic integration errors. Can. J. Chem. 78, 415 (2000).