

ABSTRACT
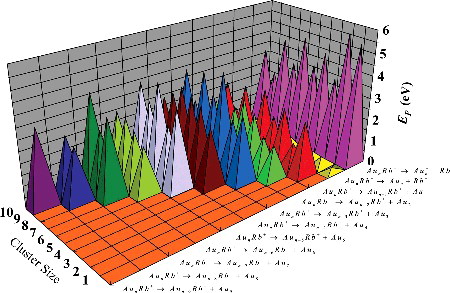
Density functional theory has been applied to study the geometric structures, relative stabilities, and electronic properties of cationic [AunRb]+ and Aun + 1+ (n = 1–10) clusters. For the lowest energy structures of [AunRb]+ clusters, the planar to three-dimensional transformation is found to occur at cluster size n = 4 and the Rb atoms prefer being located at the most highly coordinated position. The trends of the averaged atomic binding energies, fragmentation energies, second-order difference of energies, and energy gaps show pronounced even–odd alternations. It indicated that the clusters containing odd number of atoms maintain greater stability than the clusters in the vicinity. In particular, the [Au6Rb]+ clusters are the most stable isomer for [AunRb]+ clusters in the region of n = 1–10. The charges in [AunRb]+ clusters transfer from the Rb atoms to Aun host. Density of states revealed that the Au-5d, Au-5p, and Rb-4p orbitals hardly participated in bonding. In addition, it is found that the most favourable channel of the [AunRb]+ clusters is Rb+ cation ejection. The electronic localisation function (ELF) analysis of the [AunRb]+ clusters shown that strong interactions are not revealed in this study.
GRAPHICAL ABSTRACT
Disclosure statement
No potential conflict of interest was reported by the authors.