

ABSTRACT
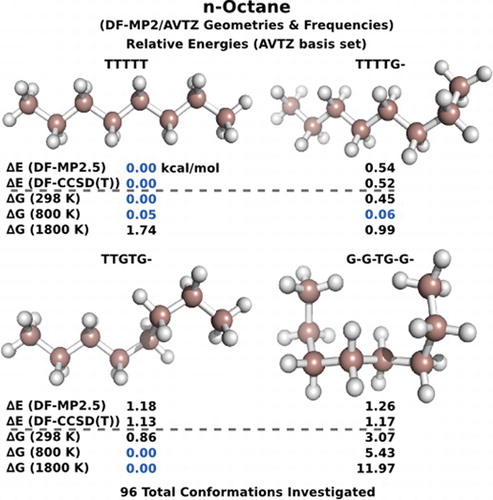
This study reports the geometries and electronic energies of n-octane's unique conformations using perturbation methods that best mimic CCSD(T) results. In total, the fully optimised minima of n-butane (2 conformations), n-pentane (4 conformations), n-hexane (12 conformations) and n-octane (96 conformations) were investigated at several different theory levels and basis sets. We find that DF-MP2.5/aug-cc-pVTZ is in very good agreement with the more expensive CCSD(T) results. At this level, we can clearly confirm the 96 stable minima which were previously found using a reparameterised density functional theory (DFT). Excellent agreement was found between their DFT results and our DF-MP2.5 perturbation results. Subsequent Gibbs free energy calculations, using scaled MP2/aug-cc-pVTZ zero-point vibrational energy and frequencies, indicate a significant temperature dependency of the relative energies, with a change in the predicted global minimum. The results of this work will be important for future computational investigations of fuel-related octane reactions and for optimisation of molecular force fields (e.g. lipids).
Acknowledgments
We would also like to thank Frank Pickard and Jadran Vrabec for helpful discussions. Rudolf Berrendorf and Javed Razzaq are gratefully acknowledged for their continuous support of the department's high-performance computing cluster.
Disclosure statement
No potential conflict of interest was reported by the authors.