
ABSTRACT
The performance of quasi-variational coupled-cluster (QV) theory applied to the calculation of activation and reaction energies has been investigated. A statistical analysis of results obtained for six different sets of reactions has been carried out, and the results have been compared to those from standard single-reference methods. In general, the QV methods lead to increased activation energies and larger absolute reaction energies compared to those obtained with traditional coupled-cluster theory.
KEYWORDS:
1. Introduction
The importance of comparing the results of a quantum chemical method to a known set of chemical properties has been long understood [Citation1]. With the G2 and G3 sets of atomisation energies and enthalpy changes, new energy functionals could be evaluated and benchmarked [Citation2]; similarly, databases of atomisation energies, equilibrium geometries and spectroscopic constants have been used to investigate the accuracy of and convergence of coupled-cluster methods with increasing basis set [Citation3,Citation4]. Statistical analysis of the properties across a set of species has supported the rigorous comparison of the performance of several ab initio methods compared to experimental values and has led to the recognition of the good performance of coupled-cluster theory with single, double and perturbative triple excitations (CCSD(T)), the deficiencies of CCSD and second-order Møller–Plesset theory (MP2), as well as understanding of the effects of orbital basis incompleteness. Statistical analysis of large collections of energies and chemical properties have become established as a standard tool through the work of Truhlar, who employed this idea to evaluate the numerous DFT functionals and determine which functional was best in predicting a general set of chemical properties [Citation5,Citation6]. This approach has culminated in the Minnesota 2.0 collection, which is comprised of computed measurements of a diverse range of chemical properties including thermochemistry, activation energies, intermolecular forces and ionisation potentials [Citation7]. Recently, this has inspired a closer examination of the performance of the Minnesota functionals with respect to a new collection of computed properties [Citation8].
In the current work, single-reference computational methods designed for representing static correlation, as well as standard approaches, have been applied to the calculation of activation and reaction energy changes for six collections of chemical reactions, to determine the accuracy and differences between the chosen methods. The intention is to determine the effectiveness of such methods for the accurate description of the reactive potential energy surface, necessary for predicting reaction kinetics and comparing mechanistic pathways.
The determination of the activation energy can potentially pose a problem for standard coupled-cluster methods such as CCSD(T). This is due to the fact that the transition state may possess multireference character because covalent bonds have been partially broken. Therefore, a simple single-reference approach may not fully capture the non-dynamic correlation effects. Over the past few years, several single-reference methods has been developed to tackle inherently multireference systems [Citation9,Citation10]. One such family of methods is quasi-variational coupled-cluster doubles, hereafter collectively denoted as the QV methods [Citation11,Citation12]. At the single and double excitation levels, standard CCSD is replaced by the ‘quasi-variational’ approximation to variational coupled cluster with double excitations, combined with variational optimisation of the energy functional with respect to variations in the reference orbitals (OQVCCD). The approach retains the N6 cost scaling of CCSD, but with an increased prefactor, because of additional matrix transformations, and additional N5 work associated with multiple integral transformations arising from the orbital variations. For full details, see [Citation11]. In previous publications, the QV methods compared favourably with multireference methods where CCSD(T) has failed dramatically [Citation13]. However, to date, they have not been applied to the determination of activation and reaction energies.
Recently, a new method in the QV family has been developed: OQVCCDAR(T), i.e. orbitally optimised QV with the asymmetric–renormalised triples correction. This method includes a more numerically robust renormalised triples approximation which can be used to produce accurate benchmarking data for the QV methods as a whole.
In the following, OQVCCDAR(T) has been used as a benchmark for five different reaction collections. The performance of several single-reference methods has been analysed relative to these benchmark calculations, with particular reference to the computation of activation and reaction energies.
2. Computational details
In order to manage and coordinate sets of computations on all of the species involved in a collection of chemical reactions, we have developed a computational framework associated with the Molpro [Citation14] quantum chemistry package. The principal entity is a database, which is a complete specification of a number of chemical reactions together with one instance of the structure, energies and other properties of every chemical species involved. The database contains the following components:
| (1) | A master file, which expresses through the XML language the definition of a number of chemical reactions, together with reference to further XML files giving the data for each chemical species. The chemical reaction is specified as a list of chemical species, each of which is assigned a stoichiometry, which is the number of times it appears on the right-hand side of the balanced chemical equation (i.e. reactants will have negative stoichiometry). Special markup can be used to tag, for example, transition states. | ||||
| (2) | For each chemical species involved in one or more of the reactions, an XML file that contains the geometry of the molecule and its calculated energy, as well as any other computed properties. This file is normally produced directly by Molpro. | ||||
| (3) | For each chemical species, a Molpro input file that will run a job with the database-contained geometry. | ||||
| (4) | A master Molpro input file, which is included by each of the molecule input files, which can be used to specify the quantum chemistry ansatz (e.g. basis set, method, density functional, etc.) that will be used for each molecule. | ||||
Both the master file and the molecule file can be validated strictly against the corresponding XML schemas. The precisely defined grammar then supports safe construction and interpretation of these files in other programs. For example, Molpro can read molecular geometry directly from the molecule XML file, and can produce a complete file that specifies geometry, basis-set and method, as well as results obtained.
We have also written several utilities for manipulating databases. The clone utility makes a copy of a database, so that it can be populated with the results of a different quantum chemistry method by running the jobs it contains, after adjusting the master input file. The analyse utility takes one or more congruent (i.e. with the same molecules and reactions, but potentially different geometries, methods and properties) databases, and evaluates the energies relative to reactants for each critical point (usually transition state and products) in each chemical reaction. In the case of more than one database, a statistical analysis is performed for the set of differences of critical-point-relative energies between each database and the first, including mean, mean absolute deviation () and maximum absolute difference (Δmax), as well as the standard deviation (Δstd) of the differences.
Two closed-shell databases were selected for the calculation of activation energies (Ea):
CRBH20 contains 20 cycloreversion transition states, the reverse processes of cycloaddition reactions. These include the fragmentation of five-membered heterocyclic rings (10 dioxazoles and 10 oxathiazoles) into cyanate and carbonyl products [Citation15]. These reactions also involve the migration of a hydrocarbon or hydrofluorocarbon substituent across a C=N bond.
BHPERI consists of 26 transition states for pericyclic reactions compiled by Goerigk and Grimme [Citation16]. These include 10 pericyclic reactions with unsaturated hydrocarbons such as an electrocyclic reaction of cyclobutene, Diels–Alder reactions with cyclopentadiene and cycloreversions of large molecules such as cis-triscyclopropacyclohexane [Citation17]. Also included are three classes of 1,3-dipolar cycloadditions, involving diazonium, nitrilium and azomethine betaines to form five-membered heterocyclic rings [Citation18]. Finally, seven Diels–Alder reactions are incorporated, involving the addition of ethylene to different five-membered heterocycles [Citation19].
Two databases were chosen to investigate solely reaction energies (ΔE):
ISOMER20. A closed-shell subset of this database was constructed from the 20 original organic isomerisation reactions [Citation20]; this now consists of reaction energies for 16 endothermic reactions. These include isomerisations of small molecules like hydrogen cyanide and isocyanic acid, and larger molecules like ketene and acetaldehyde.
DARC consists of 14 exothermic Diels–Alder reactions [Citation16,Citation21]. These include reactions of dienes like butadiene and cyclopentadiene with ethene, ethyne, maleine and maleimide.
Finally, two databases were chosen that consist of both activation and reaction energies:
O3ADD6 contains two reactions with the addition of ozone to ethene and ethyne [Citation16,Citation22]. The database is comprised of two barrier heights, two reaction energies and two van der Waals (vdW) energies for the associated ozonide complex.
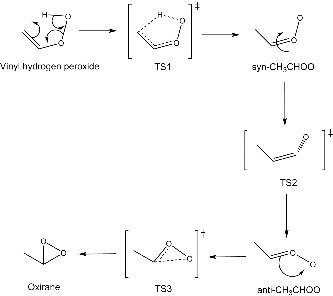
CRIEGEE is a newly constructed database which comprises a reaction pathway, as shown in , involving a Criegee intermediate. This pathway consists of three sequential transition states, and so, in total, provides a set of three activation energies and one exothermic and two endothermic reaction energies.
Figure 1. Reaction pathway which defines the CRIEGEE database.

In total, these databases contain 153 distinct chemical species that are used to calculate 51 activation energies and 37 reaction energies. Full details of the databases, including the geometries of each molecular species, are available at http:// doi.org/10.17035/d.2017.0038224181.
All six databases were evaluated with the provided geometries using the standard MP2, CCSD and CCSD(T) methods, as well as the distinguishable cluster (DCSD), quasi-variational coupled-cluster doubles with orbital optimisation (OQVCCD), with the standard perturbative correction for connected triples excitations (OQVCCD(T)), the symmetrised renormalised perturbative triples correction (OQVCCDR(T)) and OQVCCDAR(T). The mean (), standard deviation (Δstd), mean absolute deviation (
) and absolute maximum difference (Δmax) were calculated for each method relative to OQVCCDAR(T). Several different basis sets were used; however, only the largest basis set results are presented here. For the O3ADD6 database, the systems were small enough to use full coupled-cluster with single, double and triple excitations (CCSDT) as the benchmark and investigate the effects of the full inclusion of triple excitations. These calculations were carried out using Molpro's interface to the MRCC program of M. Kallay [Citation23].
3. Results and discussion
shows the activation and association energy differences compared to CCSDT for the O3ADD6 database. The first point to note is the large differences of MP2, CCSD and OQVCCD with the thermochemistry predicted by CCSDT. MP2 greatly overestimates the reaction energies of the adduct formation by 40.6 and 134.0 kJ mol−1, respectively. CCSD and OQVCCD both produce more negative reaction energies by around 30 kJ mol−1. DCSD, on the other hand, produces results for both reaction energies which are within chemical accuracy compared to CCSDT.
Table 1. Energy differences with CCSDT / kJ mol−1 for the O3ADD6 database with cc-pVDZ basis set. In the case of CCSDT, the actual energies are reported.
The non-iterative (T) correction decreases these differences substantially. For CCSD(T), the differences for the adduct reaction energies are below 1 kJ mol−1. OQVCCD(T) still predicts a more negative energy by around 5 kJ mol−1 for both reactions. The effect of the renormalisation is to decrease this quantity even more by 3.5 kJ mol−1; OQVCCDAR(T) decreases this further by 0.03 kJ mol−1.
For the vdW energies, all methods, apart from MP2, predict values close to CCSDT. DCSD excels here by producing the smallest differences. Specifically, for the ethene complex, CCSD and OQVCCD both produce larger energies, with maximum differences of 1.5 and 1.7 kJ mol−1, respectively. The effect of the standard triples correction is to reduce these quantities further. OQVCCD(T) produces more negative vdW energies than CCSD(T); these are closer to the CCSDT reference by around 0.3 kJ mol−1. The renormalisation serves to decrease these quantities again; OQVCCDAR(T) lowers this by around 0.1 kJ mol−1.
The calculation of the activation energies shows larger differences than the vdW energies. MP2 produces inconsistent results; it overestimates the first energy by 13.6 kJ mol−1 and underestimates the second by 83 kJ mol−1. CCSD and OQVCCD improve upon these results; CCSD predicts higher barrier heights of 5.5 and 5.7 kJ mol−1 for each reaction, while OQVCCD increases this difference to around 7 kJ mol−1. DCSD shows good agreement with CCSDT.
The effect of the triples correction is to lower these barrier heights below the full triples result. OQVCCD(T) predicts larger barrier heights than CCSD(T) by around 0.6 kJ mol−1 for the second reaction. Renormalisation corrects this lowering by the (T) correction and increases the barrier heights again.
The QV methods with triples corrections produce the smallest differences compared to CCSDT, with OQVCCDAR(T) differing by around 1 kJ mol−1 for both reactions. The activation energies that are calculated are in between the CCSD(T) and CCSDT results. The QV methods appear to correct for the lowering of the barrier height by the (T) correction.
The statistics for the CRBH20 database are shown in . The largest deviation occurs for MP2 with a mean difference and of 23 kJ mol−1. OQVCCD shows the next largest deviation with a mean and
of 10.4 kJ mol−1. The triples correction for the QV methods and CCSD clearly makes a large contribution to the overall activation energies. DCSD, however, shows one of the smallest deviations from OQVCCDAR(T), apart from OQVCCD(T) and OQVCCDR(T), with a mean difference of 1.98 kJ mol−1.
Table 2. Ea statistical differences (kJ mol−1) from OQVCCDAR(T) for the CRBH20 database with cc-pVTZ basis set.
Overall, including triples has the effect of lowering the activation energy. The QV methods predict higher activation energies than CCSD(T), with a mean difference approaching 3.4 kJ mol−1. The effect of the renormalised triples corrections is to increase the energy barrier. The asymmetric–renormalised triples leads to further increase, though only by around 0.01 kJ mol−1 when compared to OQVCCDR(T).
For CCSD(T), the largest individual reaction difference of 5.4 kJ mol−1 occurs for reaction 11, which involves 1,4,2-oxathiazole breaking into isothiocyanic acid and formaldehyde. However, there are no energy differences that approach 5 kJ mol−1 for the remaining nine oxathiazole rings.
The largest difference for OQVCCD of 15 kJ mol−1 occurs for reaction 3, which is an ethyl-substituted dioxazole ring. The second largest energy difference occurs for reaction 14, a fluromethyl-substituted dioxazole ring. There appears no correlation between these large energy differences and the two types of heterocyclic ring.
shows the results for the BHPERI database. Large mean differences are observed for MP2, CCSD, DCSD and OQVCCD. For this database, MP2 completely underpredicts the barrier heights by a mean of 33.9 kJ mol−1; the largest difference occurs for reaction 9 with an error of 55 kJ mol−1. OQVCCD and CCSD both show similar differences, each overpredicting the barrier height, with the largest difference also occurring for reaction 9 (a Diels–Alder reaction involving two cyclopentadienes).
Table 3. Ea statistical differences (kJ mol−1) from OQVCCDAR(T) for the BHPERI database with cc-pVTZ basis set.
The perturbative triples corrections again lead to a lowering of the barrier heights. CCSD(T) produces answers that are closer to OQVCCDAR(T), with a mean difference of −2.9 kJ mol−1. In general, the QV methods lead to an increase of the activation energies. Differences greater than 4 kJ mol−1 occur for reactions 11 and 14 which involve 1,3-dipolar cycloadditions.
The effect of using the renormalised triples formalisms is to increase the barrier heights slightly by a mean of 1.4 kJ mol−1. The asymmetric–renormalised triples leads to a further increase compared to the symmetric renormalisation. The difference between these two methods is small; the largest difference being 0.9 kJ mol−1.
presents the statistics for the reaction energies of the DARC database. Overall, these results do not show strong deviations from the OQVCCDAR(T) energies unlike the activation energies in and . Again, the largest difference occurs with MP2, which tends to underpredict the energy change with a mean difference of −13.9 kJ mol−1. The is around 23.7 kJ mol−1. DCSD shows the next largest difference after MP2, with a mean difference of 6.3 kJ mol−1. OQVCCD shows the closest match to the OQVCCDAR(T) energies, compared to the other methods without triples corrections.
Table 4. ΔE statistical differences (kJ mol−1) from OQVCCDAR(T) for the DARC database with cc-pVTZ basis set.
OQVCCDAR(T) predicts more exothermic reaction energies than all the methods, though OQVCCDR(T) produces results that are very similar; the mean difference being 0.005 kJ mol−1. CCSD(T) also shows little deviation with a mean difference of 1.6 kJ mol−1. The renormalised triples correction serves to decrease the reaction energies by about 1 kJ mol−1 compared to the standard (T) correction.
presents the statistics for a closed-shell subset of the ISOMER20 database. All the methods give a mean difference within 1 kJ mol−1of the reference values, apart from MP2. However, CCSD and OQVCCD have large Δstd values, indicating a large distribution of values that happen to cancel out each other when the mean is taken. Large absolute maximum differences for both methods occur for the isocyanic acid isomerisation to fulminic acid (reaction 8).
Table 5. ΔE statistical differences (kJ mol−1) from OQVCCDAR(T) for a subset of the ISOMER20 database with cc-pV5Z basis set.
There is also a small mean difference for DCSD; however, this is also due to a wide spread of relative results.
Overall, the QV methods with the triples predict more endothermic reaction energies than CCSD(T) and DCSD. There is little difference between the symmetric– and asymmetric–renormalisation corrections. The renormalisation does serve to slightly increase the reaction energies.
and show the statistics for the CRIEGEE database. For the activation energies, all methods have small mean differences apart from MP2, CCSD and OQVCCD. For OQVCCD, the barrier height for the first transition state differs by 16.7 kJ mol−1. It is this reaction that also produces the largest errors for CCSD. Again, DCSD produces surprisingly close results for a method without any triples correction.
Table 6. Ea statistical differences (kJ mol−1) from OQVCCDAR(T) for the CRIEGEE database with cc-pV5Z basis set.
Table 7. ΔE statistical differences (kJ mol−1) from OQVCCDAR(T) for the CRIEGEE database with cc-pV5Z basis set.
CCSD(T), OQVCCD(T) and OQVCCDR(T) all produce similar activation energies compared to OQVCDDAR(T). CCSD(T) produces a lower mean barrier height by 1.6 kJ mol−1; however, a larger difference of 3.3 kJ mol−1 is observed for the first transition state (TS1), which involves the breaking and forming of four different bonds. Compared to OQVCCD(T), the effect of the renormalised triples is to increase the activation energy in reaction 1 by 0.8 kJ mol−1 and smaller decreases for reactions 2 and 3 by 0.2 and 0.1 kJ mol−1, respectively.
presents the statistical results for the reaction energies. Again, from the mean differences, all the methods appear to be in good agreement. However, MP2 and OQVCCD show large deviations for the first and third reactions. On average, compared to CCSD(T), the QV methods produce more endothermic reaction energies for the first and second reactions, while producing a more exothermic energy for the third reaction. The largest difference for CCSD(T) again occurs for reaction 1 with a lower energy of 5.1 kJ mol−1.
4. Conclusion
There are several conclusions that can be drawn from the results obtained. First, unsurprisingly, MP2 performs poorly for the calculation of accurate activation and reaction energies. OQVCCD also does not produce satisfactorily quantitative results, especially for the calculation of activation energies. It is, therefore, essential to include the effect of connected triple excitations with OQVCCD to produce reliable results. DCSD produces excellent results for the O3ADD6 and CRBH20 databases, with differences within chemical accuracy. For the BHPERI database, the errors are larger, but are still below those of OQVCCD. For the calculation of reaction energies, OQVCCD performed better for DARC, whereas DCSD performed better with the ISOMER20 subset.
In general, the use of the QV methods leads to an increase in the activation energies and an increase in absolute reaction energies when compared to CCSD(T). From the mean differences and standard deviations, these methods produce higher barrier heights by around 2–3 kJ mol−1. However, there are individual barrier heights that CCSD(T) underestimates by 4–5 kJ mol−1. These transition states exhibit some non-dynamical correlation effects, which are, however, generally small. For the calculation of reaction energies, CCSD(T) and the QV methods are in agreement, with differences approaching 3 kJ mol−1. When compared to CCSDT, the effect of the QV methods is to correct for the limitations of the non-iterative triples and increase the barrier height. This error is again reduced with the renormalised triples corrections.
Acknowledgments
This work has been supported by EPSRC (Studentship 1375170).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- L.A. Curtiss, K. Raghavachari, P.C. Redfern, and J.A. Pople, J. Chem. Phys. 106(3), 1063–1079 (1997).
- L.A. Curtiss, K. Raghavachari, P.C. Redfern, and J.A. Pople, J. Chem. Phys. 112(17), 7374–7383 (2000).
- K.L. Bak, J. Gauss, P. Jørgensen, J. Olsen, T. Helgaker, and J.F. Stanton, J. Chem. Phys. 114(15), 6548–6556 (2001).
- F. Pawłowski, A. Halkier, P. Jørgensen, K.L. Bak, T. Helgaker, and W. Klopper, J. Chem. Phys. 118(6), 2539–2549 (2003).
- J. Zheng, Y. Zhao, and D.G. Truhlar, J. Chem. Theory Comput. 5(4), 808–821 (2009).
- P. Verma and D.G. Truhlar, Phys. Chem. Chem. Phys. 19, 12898–12912 (2017).
- R. Peverati and D.G. Truhlar, Phil. Trans. R. Soc. A 372(2011), (2014).
- N. Mardirossian and M. Head-Gordon, J. Chem. Theory Comput. 12(9), 4303–4325 (2016).
- D. Kats and F.R. Manby, J. Chem. Phys. 139(2), 021102 (2013).
- L.M.J. Huntington and M. Nooijen, J. Chem. Phys. 133(18), 184109 (2010).
- J.B. Robinson and P.J. Knowles, J. Chem. Phys. 136(5), 054114 (2012).
- J.B. Robinson and P.J. Knowles, J. Chem. Phys. 138(7), 074104 (2013).
- J.B. Robinson and P.J. Knowles, J. Chem. Theory Comput. 8(8), 2653–2660 (2012).
- H.J. Werner, P.J. Knowles, G. Knizia, F.R. Manby, and M. Schütz, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2(2), 242–253 (2012).
- L.J. Yu, F. Sarrami, R.J. O’Reilly, and A. Karton, Chem. Phys. 458, 1–8 (2015).
- L. Goerigk and S. Grimme, J. Chem. Theory Comput. 6(1), 107–126 (2010).
- V. Guner, K.S. Khuong, A.G. Leach, P.S. Lee, M.D. Bartberger, and K.N. Houk, J. Phys. Chem. A 107(51), 11445–11459 (2003).
- D.H. Ess and K.N. Houk, J. Phys. Chem. A 109(42), 9542–9553 (2005).
- T.C. Dinadayalane, R. Vijaya, A. Smitha, and G.N. Sastry, J. Phys. Chem. A 106(8), 1627–1633 (2002).
- A. Karton, S. Daon, and J.M.L. Martin, Chem. Phys. Lett. 510(4–6), 165–178 (2011).
- E.R. Johnson, P. Mori-Sánchez, A.J. Cohen, and W. Yang, J. Chem. Phys. 129(20), 204112 (2008).
- Y. Zhao, O. Tishchenko, J.R. Gour, W. Li, J.J. Lutz, P. Piecuch, and D.G. Truhlar, J. Phys. Chem. A 113(19), 5786–5799 (2009).
- Mrcc, a quantum chemical program suite written by M. Kállay, Z. Rolik, J. Csontos, P. Nagy, G. Samu, D. Mester, J. Csóka, I. Ladjánszki, L. Szegedy, B. Ladóczki, K. Petrov, M. Farkas, and B. Hégely. See also Z. Rolik, L. Szegedy, I. Ladjánszki, B. Ladóczki, and M. Kállay, J. Chem. Phys. 139, 094105 (2013), as well as: www.mrcc.hu.