
ABSTRACT
The majority of proteins perform their cellular function after folding into a specific and stable native structure. Additionally, for many proteins less compact ‘molten globule’ states have been observed. Current experimental observations show that the molten globule state can show varying degrees of compactness and solvent accessibility; the underlying molecular cause for this variation is not well understood. While the specificity of protein folding can be studied using protein lattice models, current design procedures for these models tend to generate sequences without molten globule-like behaviour. Here we alter the design process so the distance between the molten globule ensemble and the native structure can be steered; this allows us to design protein sequences with a wide range of folding pathways, and sequences with well-defined heat-induced molten globules. Simulating these sequences we find that (1) molten globule states are compact, but have less specific configurations compared to the folded state, (2) the nature of the molten globule state is highly sequence dependent, (3) both two-state and multi-state folding proteins may show heat-induced molten globule states, as observed in heat capacity curves. The varying nature of the molten globules and typical heat capacity curves associated with the transitions closely resemble experimental observations.
GRAPHICAL ABSTRACT
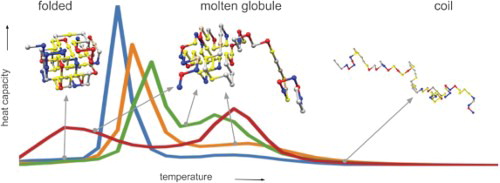
1. Introduction
One of the hallmarks of protein folding is the precision with which a specific protein sequence can fold into a well-defined topology. In the cell, the majority of proteins perform their functional role in this folded or ‘native’ structure. At high temperatures, proteins will generally unfold or denature [Citation1,Citation2]; this is due to the chain entropy becoming more dominant at high temperatures, favouring the unfolded or ‘coil’ state that is an ensemble of many extended structural configurations. The folding specificity is characterised by a high peak in the heat capacity [Citation3–8] that coincides with the heat-unfolding transition. Moreover, the transition from fully folded protein configurations to an ensemble of fully unfolded protein configurations takes place in a relatively small temperature window () [Citation4,Citation8]. Within this window, the denaturation midpoint temperature, or
in short, is the temperature at which
of the ensemble is in the folded state. Understanding the underlying biophysical pathways associated with heat capacity profiles is essential, especially since many drug development pipelines [Citation9–11] and even disease profiling techniques can be based on heat capacity scanning techniques [Citation12].
While the native protein fold is highly specific, more dynamic, relatively compact states have also been observed for many real proteins; such states are usually referred to as ‘molten globules’ [Citation13,Citation14]. These molten globules are much more compact than the fully unfolded coil state, but much less specific than the folded state; intra-chain contacts between the residues of the protein form a fluctuating ensemble [Citation14–17]. Over the last decade, it has become increasingly clear that there is a very diverse spectrum of molten globule-like states, ranging from near native compact structures (‘dry’ molten globules) to much more dynamic and more solvent accessible structures (‘wet’ molten globules) [Citation18–20]. These state ensembles also vary greatly in the extent to which their cores are solvent accessible, spanning several orders of magnitude [Citation19]. Experimentally, the denaturation of molten globule states coincides with a peak in the heat capacity, although such peaks are much smaller than those observed for unfolding [Citation21] the native structure. Note that some proteins have even been reported to be able to function in a molten globule-like state [Citation19,Citation22].
Another, very much related, discussion revolves around the folding pathway at physiological temperatures, or the free energy landscapes of proteins at temperatures where the folded state is most stable. For the folding pathway, a distinction can be made between two-state and multi-state folders [Citation23–25]. The first type of protein will follow a folding pathway from the unfolded to the folded structure, with a single barrier in the form of a transition state – note that this may be an ensemble of configurations – that separates the coil and fully folded states. Multi-state folding, with several meta-stable states between the coil and fully folded state, is associated with multiple peaks in a heat capacity versus temperature diagram [Citation8]. Typically, multi-state folding is observed for multi-domain proteins, where each domain represents a sequence–structure combination that can fold independently. Nevertheless, for single-domain proteins multi-state (un)folding pathways have also been observed [Citation25,Citation26]. In this work, we will focus on single-domain proteins.
The high specificity of protein folding and its associated thermodynamic characteristics can be captured by the classic lattice model for protein folding [Citation27]. In these models, amino acid alphabets in combination with sequence design procedures are used to generate sequence–structure combinations. This is in contrast with GO potentials, which are alternative models that effectively enforce the native structure [Citation28], but cannot capture non-specific (non-native) contacts [Citation29–31]. With sequence-based lattice models, both the peak in heat capacity and rapid transition from the folded to the coil state at increasing temperature can be replicated for a multitude of different sequence–structure combinations. Off-lattice models are able to show the same qualities in terms of specificity [Citation30,Citation32,Citation33], but parametrising such models remains a challenging task.
Computationally, molten globules have been studied using various protein simulation models, just so has the hydrophobic collapse [Citation7,Citation34–43]. However, much less is understood about the sequence–structure relationship of molten globules and how folding pathways may be influenced by the presence of such molten globule-like states. In this work, we use a simple model to answer these questions.
We adapt the design process of a sequence-based lattice model, with the goal of designing protein sequences, either with or without molten globule states. Note that we only alter the design procedure, with which we generate folding sequences, and do not modify the native structure, the protein model, the simulation model or the interaction model for these different protein sequences. We find that the folding pathways and the presence or absence of molten globule states are highly sequence dependent. Moreover, we observe that the molten globule state comes in several varieties, its characteristics determined by the sequence.
2. Methods
2.1. Folding model
Our folding model is based on the classic cubic lattice model for protein folding [Citation27,Citation43–49], along with an extension to model interactions between the protein chain and the solvent [Citation50]. In this coarse-grained model, a protein is represented by a string of amino acid beads residing on a lattice corresponding to a three-dimensional grid. Individual amino acids in the chain can interact in a pairwise manner and with an implicit solvent; the energy of such interactions is determined by statistical potentials. In our model, the internal energy of a protein chain is given by
(1) where
is the amino acid type at position i and w is the solvent. The pair potential
or
gives the interaction energies between amino acid type x and amino acid type y, or solvent w, respectively.
is the contact matrix;
is 1 if chain positions i and j are neighbouring on the lattice without being connected by a peptide bond, otherwise
is 0.
describes whether a position i in the chain is exposed to the solvent w in at least one of four possible directions:
(2) Any constants, such as the interaction energies of the pair potential
, were taken unmodified from [Citation50].
2.2. Simulation
The model was simulated by a Monte Carlo simulation algorithm using the Metropolis rule [Citation51] for trial move acceptance or rejection:
(3) where
is the Boltzmann constant, T is the simulation temperature and
is the change in system energy resulting from the proposed move. As our investigation is limited to single proteins, only internal moves are allowed. These are end moves, corner flips, crank shafts and point rotation moves [Citation47].
2.3. Design procedure
In order to generate a sequence that is able to fold specifically into a single structure, we use a design procedure in which the internal energy of a sequence is minimised, given a desired, fixed, conformation. We use a Monte Carlo based minimisation procedure largely based on existing design procedures [Citation50]. The algorithm initialises the protein chain with a random sequence. Then, iteratively, a change in amino acid type is proposed for a single residue. Acceptance of such a change (c.f. move) depends on several design criteria. The designed protein sequence needs to fold with high specificity into the given native structure. To this end, the change in internal energy difference between a fully extended conformation and the desired native confirmation is one of the optimisation criteria.
(4)
and the other energy terms are calculated using Equation Equation1
(1) . In this instance, the spatial configuration, expressed by the contact matrix C, is fixed to the native conformation and the amino acid sequence of the chain is varied, through the amino acid mapping function~
.
2.3.1. Molten globule design term
To generate a more diverse folding landscape, we include an additional objective in the design procedure. We consider the molten globule to be a set of competing compact states. To model this, we add an energy term corresponding to the change in internal energy between the native conformation and an estimation of an ensemble of compact states.
(5) Here the molten globule enthalpy
is estimated as the mean enthalpy over a small ensemble of compact structures:
(6)
(7) Here
denotes molten globule shadow state i of n. A shadow state is defined as having the native chain configuration, but with a randomly permuted amino acid sequence; this permutation order differs per shadow state but is unchanged during a single run of the design procedure.
is the design temperature; the higher the design temperature the more likely the algorithm is to accept an energetically unfavourable move. In the context of the molten globule approximation, it affects the weights of the shadow states; a lower temperature means the more energetically favourable shadow states will have a larger contribution to the total estimate
. Even at a higher design temperature, the number of shadow states n is required to be sufficiently large as to prevent a single shadow state from dominating the estimate. On a standard laptop computer, n=15 is a reasonable trade-off between accuracy and speed, although n should be set to as large a number as is computationally feasible.
In the design procedure, we also use a Monte Carlo algorithm, using the following acceptance criterion based on the sequence enthalpy:
(8) where
is the design temperature as described previously. The energy acceptance requirement is always satisfied if
; otherwise it is satisfied with probability
.
We define such that both the enthalpy difference between the folded and unfolded states, and the enthalpy difference between the folded and molten globule state, are included:
(9) To allow the molten globule contribution to this total
to be varied, a weighing parameter α is introduced. In principle, negative values of α should result in a sequence in which the internal energy difference between the native state and the molten globule state is small; conversely, positive values of α should result in a sequence in which the energy difference is large.
is the change, resulting from the proposed move, in the internal energy difference between the coil and folded states, considering the difference between the current and proposed amino acid composition of the sequence:
(10) where
is an equivalent term, but for the molten globule and folded states:
(11)
If only the energy acceptance requirement were used to constrain the moves, designed sequences would quickly converge to near-homopolymers, containing only the amino acid types with the most favourable or unfavourable interactions. A second acceptance requirement is therefore required [Citation47], enforcing realistic heterogeneity in the amino acid composition of the chain.
(12)
(13) where
is a measure of the variance of the protein chain; N is the total number of amino acids in the sequence;
is the number of occurrences in the sequence of the amino acid of type k;
is an independently controlled temperature constant called the variance temperature. Higher values of
relax the variance requirement of the chain; if set too low the majority of moves are rejected because they lower the amino acid variance; if set too high the variance requirement is not sufficiently enforced and the sequence converges to a biologically unrealistic polymer. If the variance is increased by a move (i.e. if
) it is always accepted; if the variance is lowered by a move it is accepted with probability
.
2.4. Sampling
Parallel tempering is used in order to improve the sampling within the simulation of inaccessible regions in the configurational space [Citation52]. The simulation was set to attempt 1,000,000 replica swaps in total, attempting one every 10,000 moves. Acceptance of a replica swap is governed by the acceptance rule below.
(14) In total 30 temperatures are sampled, linearly spaced on the interval
.
The heat capacity is calculated through the recorded internal energies E for every sampled configuration during a simulation. The heat capacity for a given temperature is
(15) where
is the heat capacity, E is the internal energy and T is the simulation temperature.
2.5. Order parameters
The order parameters used in the analysis of the simulation data are defined as follows. is the total number of internal contacts for a given configuration.
is the total number of internal contacts for a configuration which also exists in the native structure, or the structure that was used as the target in the design procedure.
is defined as
, or the number of internal contacts which do not exist in the native structure.
The free energy of a state i is given by
(16) where
is the Boltzmann constant, T is the temperature and
is the sampling probability of a state i, defined by one or more order parameters of choice.
2.6. Designed sequences
The parameter controlling the strength – and sign – of the molten globule minimisation objective in the design procedure, α, was varied in increments of 0.25 on the interval . Negative values of α should, in principle, increase the preference for molten globule states; positive values should decrease the preference. Ten sequences were designed for every value of α; this was done to gather more robust statistics, given the probabilistic nature of the design procedure. The length of the designed protein sequence was kept constant at 70 amino acids. The structure for which the energies were minimised was also kept constant, using a compact structure with 84 (native) contacts.
Out of the total ensemble of 90 sequences (10 replicates for 9 different values of α), we selected 5 that best illustrate variation in the folding pathways. All sequences that were used in this work are listed in Appendix. Note that not every design procedure run resulted in a sequence that would fold; negative values of α showed more extensive molten globule-like behaviour but were also less likely to fold.
3. Results
3.1. Folded, molten globule and coil state
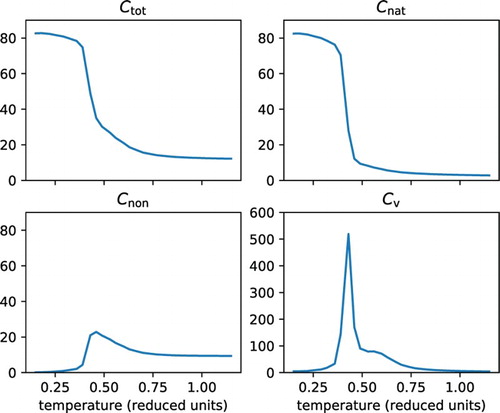
First we consider the distinct states we are able to observe for one of the designed sequences. We see that the number of native contacts () in Figure shows a very specific unfolding transition at the denaturation midpoint temperature
; this coincides with a peak in the heat capacity (
), as expected. At temperatures just above the
something interesting happens: the number of non-native contacts (
) increases sharply, while the total number of contacts (
) decreases more gradually than the number of native contacts (
). This suggests there is indeed a heat-induced molten globule state present.
Figure 1. Folding characteristics for a sequence with a heat-induced molten globule state. Simulation results are shown for a single protein sequence that was designed to fold into a specific structure with 84 native contacts. The panels show the total number of contacts (), the number of native contacts (
), the number of non-native contacts (
) and the heat capacity (
) in
, all versus the temperature in reduced units. The sharp decrease in the number of native contacts shows the transition from the folded to the molten globule state, associated with the high peak in the heat capacity curve. The transition from the heat-induced molten globule state to the coil state can be most easily seen by the decrease in the number of non-native contacts, associated with the shoulder – or very shallow peak – in the heat capacity curve.
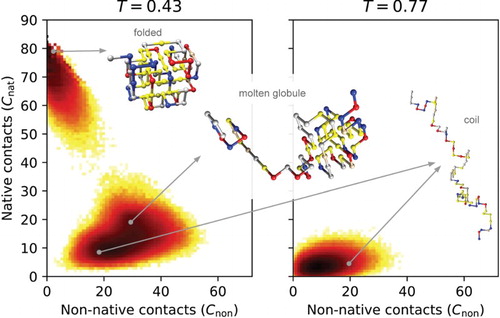
This molten globule state is much more apparent if we consider the two-dimensional free energy landscape using and
as order parameters, as shown in Figure (T=0.43) for the same sequence. Around the
there are indeed three accessible states present: the fully folded state with
, the molten globule state with
and
, and finally the unfolded coil state with
and
. At higher temperatures (
), this molten globule state gradually disappears, only leaving the coil state with highly extended conformations; this coils state is shown in Figure (T=0.77).
Figure 2. The folded, molten globule and coil states. Two-dimensional free energy landscapes derived from lattice model simulations are shown at two different temperatures for the same protein sequence as shown in Figure ; native contacts are shown on the Y -axis, non-native contacts
on the X-axis. At T=0.43 the native state, the molten globule state and coil state are all populated. Note that
for this protein sequence. At the higher temperature, T=0.77, the coil state clearly dominates the configurational ensemble. The lattice model configurations show hydrophobic residues in yellow, positively charged in blue, negatively charged in red and polar residues in grey.
3.2. The molten globule transition is gradual
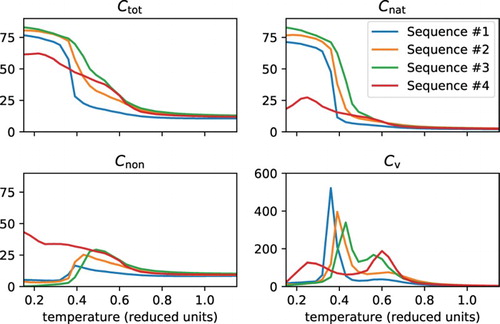
We can also observe that the molten globule state has characteristics very distinct from the fully folded state. Generally, the molten globule state is a more gradual, much less specific state. This becomes most apparent when we consider the ensemble characteristics of this state under changing temperature.
The transition from the molten globule state to the coil state is characterised by a more gradual decrease in the total number of internal contacts, compared to the unfolding transition (Figures and , panel ). The less specific character of the molten globule state, compared to the folded state, is also apparent in the lower heat capacity peak at the transition to coil (Figures and , panel
), compared to the unfolding transition.
Figure 3. Folding characteristics for various protein sequences. Simulation results are shown for various protein sequences all designed to fold into the same structure with 84 native contacts. The panels show the total number of contacts (), the number of native contacts (
), the number of non-native contacts (
) and the heat capacity (
) in
, all versus the temperature in reduced units. Sequences #1, #2 and #3 fold into the native state, while sequence #4 does not, as evidenced by the lack of a peak in the heat capacity plot and the low number of native contacts at low temperatures. The ensemble characteristics of the molten globule states, and of the associated transition states, are highly variable and sequence dependent.
Finally, we are able to observe that the nature of the molten globule state, in terms of the order parameters, slowly changes with temperature, just as observed for the coil state. Comparing the two temperatures in Figure , we can see that the number of contacts (native and non-native) declines as the temperature increases. In contrast, the folded state remains most stable around a very specific structure, characterised by , for all folding sequences.
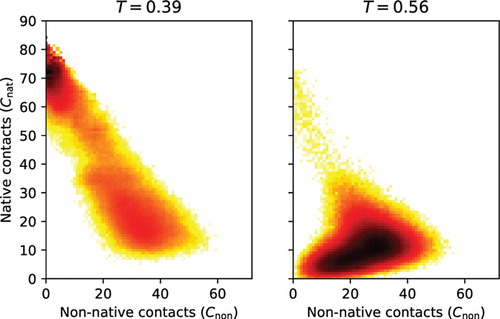
Figure 4. Free energy landscape of a multi-state folder. Two-dimensional free energy landscapes derived from simulations are shown at two different temperatures for the same protein sequence #3 used in Figure 3; native contacts are shown on the Y -axis, non-native contacts
on the X-axis. At T=0.39, the protein is most stable in its native structure, but competing molten globule-like states are also sampled. At T=0.56, the protein is transitioning from the molten globule state to a non-compact coiled state.
3.3. Sequence dependence of molten globule states
More generally, Figure shows that the nature of the molten globule state is very much sequence dependent. We designed several different sequences for the same native structure. Each sequence shows a different type of molten globule state, with a varying number of total and native contacts; moreover, the temperature at which the different sequences show the transition from molten globule to coil, as well as the height of the associated peak in the heat capacity, is strongly dependent on the sequence. In fact, sequence #4, as depicted in Figure , does not even fold into the designed structure – or in any other structure. Nevertheless it shows two distinct state transitions, the first changing between two types of molten globule states, the second changing between the molten globule and the coil state.
The unfolding temperature also shows a dependence on the sequence. Note that this is also observed for real proteins with a similar structure; for example, it is possible to engineer proteins to become more thermally stable [Citation53].
For some sequences – but not all – the molten globule state is already present at temperatures at which the folded state is most stable (i.e. for ), as shown in Figure . Here the molten globule state forms a competing state to the folded state. This implies that the folding pathway, at a physiological temperature, should visit a folding intermediate; and hence we would observe a single-domain multi-state folder. Other designed proteins show that, when a molten globule is visited in the heat unfolding pathway, there is not necessarily such a meta-stable state present at temperatures below
. Hence not all sequences with a heat-induced molten globule state are multi-state folders.
4. Discussion
The use of a modified sequence design procedure allowed us to generate several protein sequences for a single protein structure with different folding pathways, as well as different types of molten globule states. In the foremost place, these results show that folding pathways and the existence of molten globule states are highly sequence dependent; the molten globule state is not intrinsically tied to the native structure, nor does it show high specificity for a particular conformation. In fact, we show that even protein sequences without any specificity for a native structure, i.e. natively disordered proteins, can exhibit (multiple) molten globule-like states. Note that experimentally similar temperature-dependent changes in the compactness of intrinsically disordered protein regions have been observed [Citation54].
The molten globule states show peaks in the heat capacity curve, albeit much less high than those observed for the folding transition. Moreover, the temperature at which the molten globule states are present is highly variable, sometimes coinciding with the range in which the protein is stably folded, thereby effectively introducing a competing meta-stable state. Note that these results very much agree with current experimental findings that molten globules are very heterogeneous [Citation19]; moreover, different types of molten globules have been observed for many different proteins [Citation19]. Lastly, shoulders and double peaks in the heat capacity have also been observed in Differential Scanning Calorimetry (DSC) experiments for real proteins [Citation8].
While the adapted design procedure did allow for the design of a rich landscape of sequences with different molten globule-like states, the controlling design parameter, α, did not have a unique correspondence to specific molten globule-like features of the designed protein sequence. Generally, negative values of the α parameter resulted in sequences exhibiting more compact molten globule-like states. A negative α also adversely impacted the success rate of folding into a unique structure. Conversely, positive values of α in the design procedure did not consistently produce sequences with a less compact or destabilised molten globule state. In general, the variability of folding characteristics between sequences designed with the same α was large; this, together with the inherent stochasticity of the design algorithm, makes it difficult to steer the design procedure towards a specific type of molten globule state. For this study, more control over the design procedure was not necessary, as we obtained our desired variation in folding pathways. Nevertheless, the design procedure does allow for some control over the appearance and characteristics of molten globule-like states.
It may not be unrealistic to develop a similar design procedure for real proteins. While the protein structure prediction problem remains an unsolved scientific challenge [Citation55], the challenge of designing a sequence that folds into a specific structure has been tackled much more successfully. In fact, the most successful computational design algorithm for real protein structures [Citation56] resembles in essence the simple design procedure for the lattice model [Citation47,Citation50], with the addition that small structural changes are also allowed.
Our results indicate that the absence or presence of molten globule states strongly depend on the sequence. Hence, it may be feasible to engineer targeted mutations in an existing protein sequence to drive it away from or towards a sequence capable of forming molten globule-like states. Moreover, we show that similar folds can have different folding pathways, suggesting that it is possible to alter folding pathways for a given protein structure by redesigning its sequence. In particular, destabilising a competing molten globule state may be of interest: molten globule-like states can lead to irreversible unfolding [Citation8] and potentially to aggregation in real systems.
Lastly, understanding the nature of thermodynamic characteristics, including competing states in the folding pathways, is extremely important for understanding heat capacity curves that are used in a large variety of applications, including disease profiling [Citation12] and drug design [Citation9–11].
Acknowledgements
We would like to thank Peter Bolhuis for asking a question that triggered us to write up this work and Peter Crowe for carefully proofreading this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
S. Abeln http://orcid.org/0000-0002-2779-7174
Additional information
Funding
References
- N.N. Khechinashvili, J. Janin, and F. Rodier, Protein Sci. 4 (7), 1315 (1995). doi: 10.1002/pro.5560040707
- R.L. Baldwin, J. Mol. Biol. 371 (2), 283 (2007). doi: 10.1016/j.jmb.2007.05.078
- P.L. Privalov, E.I. Tiktopulo, S.Y. Venyaminov, Y.V. Griko, G.I. Makhatadze, and N.N. Khechinashvili, J. Mol. Biol. 205 (4), 737 (1989). doi: 10.1016/0022-2836(89)90318-5
- G. Castronuovo, Thermochim. Acta 193, 363 (1991). doi: 10.1016/0040-6031(91)80193-M
- A.N. Naganathan, J.M. Sanchez-Ruiz, and V. Muñoz, J. Am. Chem. Soc. 127 (51), 17970 (2005). doi: 10.1021/ja055996y
- N.V. Prabhu and K.A. Sharp, Annu. Rev. Phys. Chem. 56, 521 (2005). doi: 10.1146/annurev.physchem.56.092503.141202
- S. Abeln, M. Vendruscolo, C.M. Dobson, and D. Frenkel, PLoS ONE 9 (1), e85185 (2014). doi: 10.1371/journal.pone.0085185
- S. Mazurenko, A. Kunka, K. Beerens, C.M. Johnson, J. Damborsky, and Z. Prokop, Sci. Rep. 7 (1), 16321 (2017). doi: 10.1038/s41598-017-16360-y
- A. Velazquez-Campoy, S.A. Leavitt and E. Freire, Protein–Protein Interact. Methods Appl., (Humana Press, New Jersey, 2004), Vol. 261, pp. 35–54.
- K. Vuignier, J. Schappler, J.L. Veuthey, P.A. Carrupt, and S. Martel, Anal. Bioanal. Chem. 398 (1), 53 (2010). doi: 10.1007/s00216-010-3737-1
- E. Prenner and M. Chiu, J. Pharm. Bioallied Sci. 3 (1), 39 (2011). doi: 10.4103/0975-7406.76463
- S. Vega, M.A. Garcia-Gonzalez, A. Lanas, A. Velazquez-Campoy, and O. Abian, Sci. Rep. 5 (1), 7988 (2015). doi: 10.1038/srep07988
- M. Ohgushi and A. Wada, FEBS Lett. 164 (1), 21 (1983). doi: 10.1016/0014-5793(83)80010-6
- D.N. Brems and H.A. Havel, Proteins 5 (1), 93 (1989). doi: 10.1002/prot.340050110
- P. Jennings and P. Wright, Science (80) 262 (5135), 892 (1993). doi: 10.1126/science.8235610
- A.K. Dunker, J.D. Lawson, C.J. Brown, R.M. Williams, P. Romero, J.S. Oh, C.J. Oldfield, A.M. Campen, C.M. Ratliff, K.W. Hipps, J. Ausio, M.S. Nissen, R. Reeves, C. Kang, C.R. Kissinger, R.W. Bailey, M.D. Griswold, W. Chiu, E.C. Garner, and Z. Obradovic, J. Mol. Graph. Model 19 (1), 26 (2001). doi: 10.1016/S1093-3263(00)00138-8
- D. Eliezer, P.A. Jennings, H.J. Dyson, and P.E. Wright, FEBS Lett. 417 (1), 92 (1997). doi: 10.1016/S0014-5793(97)01256-8
- V.N. Uversky and O.B. Ptitsyn, J. Mol. Biol. 255 (1), 215 (1996). doi: 10.1006/jmbi.1996.0018
- R.L. Baldwin and G.D. Rose, Curr. Opin. Struct. Biol. 23 (1), 4 (2013). doi: 10.1016/j.sbi.2012.11.004
- V.N. Uversky, Biotechnol. J. 10 (3), 356 (2015). doi: 10.1002/biot.201400374
- I. Nishii, M. Kataoka, and Y. Goto, J. Mol. Biol. 250 (2), 223 (1995). doi: 10.1006/jmbi.1995.0373
- K. Vamvaca, I. Jelesarov, and D. Hilvert, J. Mol. Biol. 382 (4), 971 (2008). doi: 10.1016/j.jmb.2008.07.049
- Y. Bai and S.W. Englander, Proteins Struct. Funct. Genet. 24 (2), 145 (1996). doi: 10.1002/(SICI)1097-0134(199602)24:2<145::AID-PROT1>3.0.CO;2-I
- Y.J. Tan, M. Oliveberg, and A.R. Fersht, J. Mol. Biol. 264 (2), 377 (1996). doi: 10.1006/jmbi.1996.0647
- I.E. Sánchez and T. Kiefhaber, J. Mol. Biol. 325 (2), 367 (2003). doi: 10.1016/S0022-2836(02)01230-5
- M. Baclayon, P. van Ulsen, H. Mouhib, M.H. Shabestari, T. Verzijden, S. Abeln, W.H. Roos, and G.J. Wuite, ACS Nano 10 (6), 5710 (2016). < DOI: 10.1021/pacsnano.5b07072>.
- A. Sali, E. Shakhnovich, and M. Karplus, J. Mol. Biol. 235 (5), 1614 (1994). doi: 10.1006/jmbi.1994.1110
- H. Taketomi, Y. Ueda, and N. Gō, Chem. Biol. Drug. Des. 7 (6), 445 (1975).
- N. Combe and D. Frenkel, J. Chem. Phys. 118 (19), 9015 (2003). doi: 10.1063/1.1567256
- I. Coluzza, Mol. Phys. 113 (17–18), 2905 (2015). doi: 10.1080/00268976.2015.1043031
- I. Coluzza, J. Phys. Condens. Matter 29 (14), 143001 (2017). doi: 10.1088/1361-648X/aa5c76
- I. Coluzza, PLoS ONE 9 (12), e112852 (2014). doi: 10.1371/journal.pone.0112852
- V. Bianco, G. Franzese, C. Dellago, and I. Coluzza, Phys. Rev. X 7 (2), 021047 (2017).
- V. Daggett and M. Levitt, Proc. Natl. Acad. Sci. U.S.A. 89 (11), 5142 (1992). doi: 10.1073/pnas.89.11.5142
- A.E. Mark and W.F. van Gunsteren, Biochemistry 31 (34), 7745 (1992). doi: 10.1021/bi00149a001
- L.J. Smith, C.M. Dobson, and W.F. Van Gunsteren, J. Mol. Biol. 286 (5), 1567 (1999). doi: 10.1006/jmbi.1999.2545
- P.R. ten Wolde and D. Chandler, Proc. Natl. Acad. Sci. 99 (10), 6539 (2002). doi: 10.1073/pnas.052153299
- H. Schäfer, L.J. Smith, A.E. Mark, and W.F. Van Gunsteren, Proteins Struct. Funct. Genet. 46 (2), 215 (2002). doi: 10.1002/prot.1166
- W.B. Hu and D. Frenkel, J. Phys. Chem. B 110 (8), 3734 (2006). doi: 10.1021/jp056679g
- K. Singhal, J. Vreede, A. Mashaghi, S.J. Tans, and P.G. Bolhuis, PLoS Comput. Biol. 11 (10), e1004444 (2015). doi: 10.1371/journal.pcbi.1004444
- P. Kukic, A. Kannan, M.J.J. Dijkstra, S. Abeln, C. Camilloni, and M. Vendruscolo, PLOS Comput. Biol. 11 (10), e1004435 (2015). doi: 10.1371/journal.pcbi.1004435
- C. Cardelli, V. Bianco, L. Rovigatti, F. Nerattini, L. Tubiana, C. Dellago, and I. Coluzza, Sci. Rep. 7 (1), 4986 (2017). doi: 10.1038/s41598-017-04720-7
- E. van Dijk, P. Varilly, T.P.J. Knowles, D. Frenkel, and S. Abeln, Phys. Rev. Lett. 116 (7), 078101 (2016). doi: 10.1103/PhysRevLett.116.078101
- E.I. Shakhnovich and A.M. Gutin, Proc. Natl. Acad. Sci. 90 (15), 7195 (1993). doi: 10.1073/pnas.90.15.7195
- E.I. Shakhnovich and A.M. Gutin, Protein Eng. 6 (8), 793 (1993). doi: 10.1093/protein/6.8.793
- E.I. Shakhnovich, Phys. Rev. Lett. 72, 3907 (1994). doi: 10.1103/PhysRevLett.72.3907
- I. Coluzza, H.G. Muller, and D. Frenkel, Phys. Rev. E. Stat. Nonlin. Soft. Matter. Phys. 68 (4 Pt 2), 46703 (2003). doi: 10.1103/PhysRevE.68.046703
- I. Coluzza and D. Frenkel, Phys. Rev. E. Stat. Nonlin. Soft. Matter. Phys. 70 (5 Pt 1), 51917 (2004). doi: 10.1103/PhysRevE.70.051917
- I. Coluzza and D. Frenkel, Biophys. J 92 (4), 1150 (2007). doi: 10.1529/biophysj.106.084236
- S. Abeln and D. Frenkel, Biophys. J. 100 (3), 693 (2011). doi: 10.1016/j.bpj.2010.11.088
- N. Metropolis, A.W. Rosenbluth, M.N. Rosenbluth, A.H. Teller, and E. Teller, J. Chem. Phys. 21, 1087 (1953). doi: 10.1063/1.1699114
- I. Coluzza and D. Frenkel, Chem. Phys. Chem. 6 (9), 1779 (2005). doi: 10.1002/cphc.200400629
- F. Pucci and M. Rooman, Curr. Opin. Struct. Biol. 42, 117 (2017). doi: 10.1016/j.sbi.2016.12.007
- H. Sánchez, M.W. Paul, M. Grosbart, S.E. Van Rossum-Fikkert, J.H. Lebbink, R. Kanaar, A.B. Houtsmuller, and C. Wyman, Nucleic Acids Res. 45 (8), 4507 (2017). doi: 10.1093/nar/gkx084
- J. Moult, K. Fidelis, A. Kryshtafovych, T. Schwede, and A. Tramontano, Proteins Struct Funct Bioinform. 84 (S1), 4 (2016).
- D. Gront, D.W. Kulp, R.M. Vernon, C.E.M. Strauss, and D. Baker, PLoS ONE 6 (8), e23294 (2011). doi: 10.1371/journal.pone.0023294
Appendix. Sequences of designed proteins in FASTA format
