ABSTRACT
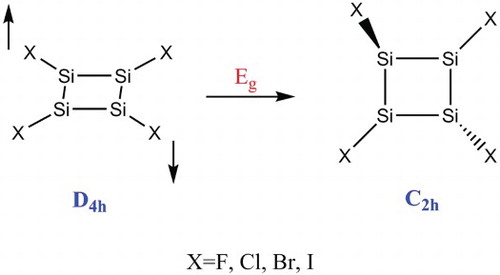
The variability of planar rings in Si4X4 (X = F, Cl, Br, I) molecules caused by the pseudo-Jahn–Teller impact (PJTE) was evaluated as an original PJTE work. Optimisation and the following frequency calculations in these molecules illustrated that in high-symmetry planar (with D4h symmetry) geometry, all of these compounds were unstable and their structures were puckered to lower C2h symmetry stable geometry. Furthermore, the vibronic coupling interaction between 1A1g ground and the first 1Eg excited states through (1A1g + 1Eg) ⊗ eg PJTE problem was the cause of non-planarity of the four-member ring and the symmetry breaking phenomenon in those series. The calculated gaps (Δ) between the ground state and the Eg excited state, the vibronic coupling (F) and ground state primary force constant values (k1) were obtained from the numerical fitting of the ground state adiabatic potential energy surface with the analytical expressions of these molecules. Finally, natural bond analysis (NBO) was used for the design of the strongest interaction and natural atomic charges of these structures.
GRAPHICAL ABSTRACT