

ABSTRACT
Potential energy surfaces and molecular dynamics of the intramolecular 1, 3-dipolar cycloaddition and ene reaction of a nitrile oxide with an alkene were performed in the gas phase and in dichloromethane with density functional theory. One hundred trajectories were propagated in the gas phase and in dichloromethane, respectively. Twenty percent of the trajectories in the gas phase involve bicyclic intermediate and the mean time gap is 472fs. A dynamically stepwise reaction is observed. In dichloromethane, more reactive trajectories were obtained and the time gap is larger than that in the gas phase.
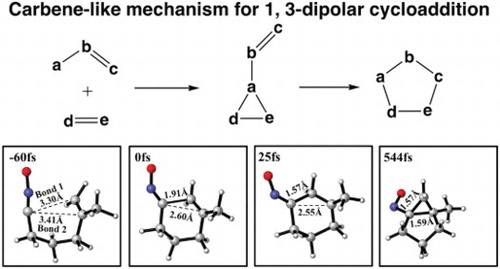
GRAPHICAL ABSTRACT
Acknowledgment
We are grateful to the National Science Foundation (NSF) (CHE-1361104) for financial support. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF (OCI-1053575). Y. Y also thanks Beijing municipal high level innovative team building programme (IDHT20180504) and China Scholarship Council (CSC) for financial support of this research.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Zhongyue Yang http://orcid.org/0000-0003-0395-6617
Notes
* Dedicated to the memory of the great theoretician and friend, Dieter Cremer.