

ABSTRACT
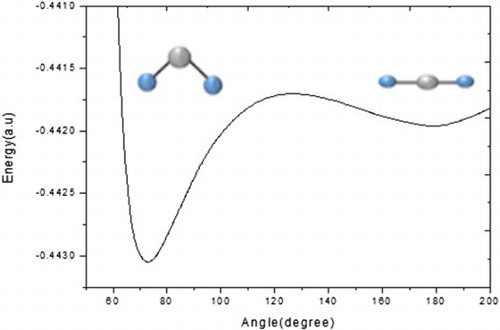
We present a theoretical study of the ground electronic state potential of the Ca+Ar2 complex and of its photoabsorption spectra, simulated at temperatures ranging between 20 and 220 K. These calculations exploit a Monte-Carlo (MC) method, based on a one-electron pseudo-potential approach. A pairwise additive potential fitted to coupled cluster ab initio points, is used to model the Ca+Ar2 complex. Our study shows that the most stable form of Ca+Ar2 is a bent C2v structure, whereas the linear isomer is located at around 90 ± 10 cm−1 above in energy. The analysis of the photoabsorption spectra establishes that a structural transition from bent Ca+Ar2 to linear ArCa+Ar occurs at T∼100 K. Trends in binding energies of both isomers, bond lengths and bond angles are also discussed. Molecular orbital overlaps provide an explanation for the order of stability between the bent and linear structures.
GRAPHICAL ABSTRACT
Acknowledgments
We would like to thank Dr. F. Spiegelman for fruitful discussions. We gratefully acknowledge the support of the COST Action CM1405 entitled MOLIM: Molecules in Motion.
Disclosure statement
No potential conflict of interest was reported by the authors.