
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Assessing the permeation rate of drug-like molecules across a lipid membrane is of paramount importance for pharmaceutical applications. While the Meyer-Overton rule relates the permeability coefficient to a few thermodynamic properties, its accuracy is limited by the homogeneity assumption. In this work, we extract an analogous relation for the permeation of a small solute through a lipid bilayer. This is obtained by systematically screening a subset of chemical space by means of high-throughput coarse-grained simulations, and relying on the accurate inhomogeneous solubility-diffusion model (ISDM). We connect the permeability coefficient of a compound to two molecular descriptors: partitioning free-energy and acid dissociation constant. In the ISDM the permeability is a functional of the potential of mean force. We discuss how – and when – combining together these profiles for a large variety of compounds can result in a smooth dependence of the permeability on the molecular descriptors. We focus on acidic molecules: for weak acids, the permeability largely depends on the main free-energy differences of the problem, which directly link to our molecular descriptors. For strong acids, the multivalued nature of the permeability requires to introduce a third variable, characterising the amphiphilicity of the small molecule.
Abbreviations: CG: coarse-grained; HTCG: high-throughput coarse-grained; PMF: potential of mean force; ISDM: inhomogeneous solubility-diffusion model; DOPC: 1; 2-Dioleoyl-sn-glycero-3-phosphocholine
GRAPHICAL ABSTRACT
1. Introduction
The strong binding affinity of a small drug-like candidate to the specific intracellular target, alone, does not guarantee its efficacy. In addition to toxicity issues, the efficacy can be severely compromised by the presence of prohibitively large time scales for the compound to cross the lipid barrier enclosing the cell environment [Citation1]. Understanding the transport mechanism of small molecules through lipid bilayers is therefore of fundamental importance for pharmaceutical applications.
Transmembrane transport processes are usually divided in two classes [Citation2]: (i) carrier-mediated processes, and (ii) passive permeation, i.e. diffusion of the permeant driven only by its concentration gradient and thermal fluctuations. Although both classes can contribute to the total transport of a solute, passive permeation is believed to play a dominant role in the case of small drug-like molecules [Citation3]. The permeability coefficient, P, quantifies the rate of passive permeation of a compound across the membrane. P is defined as [Citation4]
(1)
(1) where J is the steady-state flux per unit area of the solute arising from its concentration difference
between the two water compartments adjacent to the membrane. Equation (Equation1
(1)
(1) ) makes it apparent that a compound characterised by a low permeability coefficient will experience more difficulties in crossing the lipid membrane, in this way reducing the amount of substance delivered to the intended intracellular target.
Pharmacokinetic issues associated to insufficient permeation are among the major causes of high attrition rates in drug discovery [Citation1]. Given the complexity of in vivo experiments, several in vitro methods have been developed to systematically estimate the permeability coefficient of small molecules, in order to screen them at early stages in the discovery process [Citation5–7]. These systems have contributed to the expansion of permeability databases due to the possibility of performing high-throughput experiments. At the same time, they typically provide limited insight into the microscopic origin of permeation.
From a theoretical point of view, the first model of solute transport across a lipid bilayer is the Meyer-Overton rule [Citation8,Citation9]. It approximates the membrane as a homogeneous slab, for which an application of Fick's first law of diffusion provides
(2)
(2) In Equation (Equation2
(2)
(2) ), σ is the thickness of the bilayer core, while D and K are the diffusivity and partitioning coefficient of the compound, respectively. In practice, K is usually further approximated by the partitioning coefficient between water and a bulk apolar solvent (e.g. octanol).
Despite its simplicity, the Meyer-Overton rule identifies key properties associated with the permeability: due to the dependence on the partitioning coefficient, apolar compounds are characterised by higher permeation rates [Citation10]. However, its main limitation stems from the homogeneity assumption, at odds with the inherent asymmetry of a lipid bilayer. Accounting for the heterogeneity of the medium results in the inhomogenous solubility-diffusion model (ISDM) [Citation11,Citation12], which describes the diffusion process in terms of a one-dimensional Smoluchowsky equation along z, the normal distance of the compound from the bilayer midplane [Citation13]. From the ISDM, the permeability coefficient of a compound can be calculated as an integral over the membrane extension of its potential of mean force (PMF), , and local diffusivity,
, as
(3)
(3) with
. Equations (Equation2
(2)
(2) ) and (Equation3
(3)
(3) ) become equivalent only in the case of a flat potential of mean force and constant diffusivity – i.e. if homogeneity holds. The ISDM paved the way for an in silico estimation of permeability coefficients, where
and
are determined by means of enhanced-sampling classical molecular dynamics simulations [Citation13–19]. Predictions were shown to exhibit extremely high correlation with either in vitro or in vivo permeability measurements [Citation15,Citation20], making in silico methods a robust, complementary approach to experimental investigations. Unfortunately, the extensive computational resources required for convergence of the atomistic conformational sampling – roughly
CPU hours per chemical compound – have so far limited these calculations to few small molecules with very specific chemistries, see e.g. Ref. [Citation15,Citation20].
The importance of the Meyer-Overton rule lied in its ability to relate the permeability of a compound to a key molecular descriptor directly linked to the thermodynamics of the process. However, the rule has been recently shown to overestimate the permeabilities calculated through the ISDM, which were observed to be nonlinear in the partitioning coefficient [Citation19]. Moreover, in contrast to the ISDM the Meyer-Overton rule neglects the impact of the protonation state of a compound on its permeation rate [Citation15].
Given the accuracy of the ISDM, being able to connect its predictions to a set of simple thermodynamic properties would be of fundamental importance to obtain rapid estimates of permeability coefficients. Equation (Equation3(3)
(3) ) highlights that P is a functional of the diffusivity and potential of mean force. For small molecules
was shown to be largely insensitive to the chemical detail [Citation15], so that the permeation rate is mainly affected by variations in the PMF. Extracting the link between thermodynamics and permeability in the ISDM would require to extensively probe at least a representative subset of the spectrum of possible drug-like molecules, to systematically investigate the impact of the PMF on the permeability coefficient. The computational resources associated to PMF calculations at an atomistic resolution, combined with the estimated size of drug chemical space –
compounds [Citation21] – make this approach unfeasible in practice.
The overwhelming number of possible compounds thus calls for the introduction of new strategies that allow to more comprehensively cover large regions of chemical space. In this perspective, we recently proposed the use of coarse-grained (CG) models as a powerful instrument to preserve accurate thermodynamics while ‘clustering’ chemical space, in this way enabling a more systematic and efficient in silico exploration of chemical diversity [Citation22,Citation23].
In CG models, subsets of atoms are grouped together into elementary units (beads), with interactions tuned to capture a set of structural and/or thermodynamic properties of the original system [Citation24]. In the Martini CG force field, the fundamental building blocks are a few bead types that aim at reproducing the partitioning free energy of small organic fragments between solvents of different polarity [Citation25,Citation26]. Due to the nature of the properties chosen as target, Martini proved to be very successful in reproducing the thermodynamics of insertion of small molecules in lipid bilayers [Citation27,Citation28], at the same time significantly reducing the computational investment [Citation29].
The finite number of interaction types in the model is such that many chemically different small molecules with similar shape and polarity are mapped onto the same CG representation [Citation22]. In contrast to traditional in silico methods at an atomistic resolution, the small number of bead types further enables a more systematic exploration of the possible variations in the thermodynamics of a small molecule: this can be obtained by combinatorially constructing all coarse-grained compounds up to a certain size (Figure (a)).
Figure 1. (a): we employ high-throughput coarse-grained simulations to screen a representative subset of the chemical space of small organic molecules. The systematic insertion of this large variety of compounds with very different polarities in a lipid bilayer results in an ensemble of PMFs, which dictate their permeability coefficient. (b): we highlight PMFs from the two main classes of compounds analyzed in this work: polar (top) and apolar (bottom). (c): Construction of the effective PMF (yellow area) contributing to the total resistivity
starting from the neutral (red) and charged (blue) PMFs of a compound. We highlight the main free-energy differences of the problem: the water membrane free-energy
of the neutral compound, and the acid/base free energy difference
dictating the shift between the two PMFs, directly linked to the ap
.
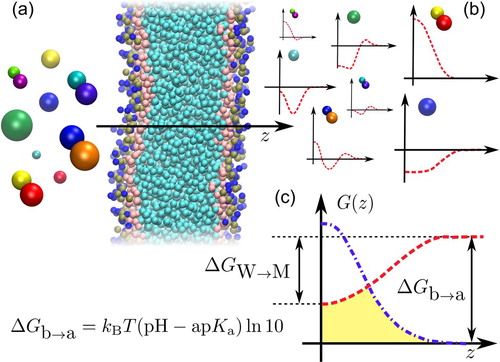
We recently took advantage of these features to shed new light on the thermodynamics of insertion of a small solute in a lipid membrane. In Ref. [Citation22] we employed high-throughput coarse-grained (HTCG) simulations to sample a representative subset of the chemical space of small organic molecules in the range 30−160 Da. Our comprehensive investigation highlighted that the overall shape of can be reconstructed only from the partitioning free energy
– i.e. the free-energy difference of transferring the solute from bulk water to the bilayer midplane, see Figure (c).
In Ref. [Citation23], by feeding HTCG results into Equation (Equation3(3)
(3) ) and accounting for the possible protonation states of a compound we identified a smooth relation expressing the permeability coefficient as a function of two molecular descriptors:
and acid dissociation constant, p
. This enabled permeability predictions for an unprecedented 500,000 small molecules including acidic, basic, and zwitterionic compounds, whose accuracy was validated against experimental as well as atomistic simulation results.
Importantly, the relation in Ref. [Citation23] connects the permeation rate to only two thermodynamic properties, providing an alternative to the Meyer-Overton rule for predicting permeability coefficients in the framework of the accurate ISDM. Results were obtained by combining together permeabilities for compounds with highly different polarities, characterised by a large variety of neutral and charged potentials of mean force (Figure (a)). In this work, we analyze in more detail the impact of the molecular descriptors on the permeation rate, resulting – when possible – in a smooth dependence of the permeability coefficient on these thermodynamic variables. We here focus on acidic compounds, described by the acidic p or ap
, separately discussing neutral compounds (
), weak acids (
) and strong acids (
). For neutral compounds, the permeability is largely insensitive to the detailed shape of the underlying PMF, but mainly dictated by
(Figure (c)) [Citation16]. This free-energy difference is not correctly accounted for in the Meyer-Overton rule, thus generating the observed discrepancies with ISDM predictions [Citation19]. Moving towards weak acids, the ap
comes into play. However, the insensitivity of the permeability on the shape of the PMF suggests that the permeation rate mainly depends on a combination of two free-energy differences:
and
, where the latter is the free energy of neutralising the compound in bulk water, directly linked to the ap
(Figure (c)). This results in the smoothness of the permeability coefficient observed as a function of our thermodynamic variables. Finally, for strong acids, at fixed ap
the permeability coefficient does no longer smoothly depend on
. This requires to introduce a third variable characterising the degree of amphiphilicity of the small molecule. In this case, our results provide a lower bound for the permeation rate of a compound: at a fixed ap
, the permeability coefficient cannot be lower than a value dictated by the combination of the two free-energy differences,
and
.
2. Methods
2.1. Molecular dynamics simulations
Molecular dynamics simulations of the Martini force field [Citation25,Citation26,Citation30] were performed in Gromacs 4.6.6 [Citation31]. We employed an integration timestep , where τ is the model's natural unit of time. Control over the system temperature (
) and pressure (
) was obtained by means of a Parrinello-Rahman barostat [Citation32] and a stochastic velocity rescaling thermostat [Citation33], with coupling constant
and
respectively. A membrane of
containing N=128 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipids (64 per layer),
water molecules,
antifreeze particles [Citation26] and enough counterions to ensure charge neutrality was generated by means of the Insane building tool [Citation34], and subsequently minimised, heated up, and equilibrated.
We determined potentials of mean force by means of umbrella sampling simulations [Citation35]. For each analyzed compound, we employed 24 windows with harmonic biasing potentials (
) centred every
along the normal to the bilayer midplane. In each window, two solute molecules were placed in the membrane in order to increase sampling and alleviate leaflet-area asymmetry [Citation36,Citation37]. Each production simulation was run for
, and potentials of mean force were extracted by means of the weighted histogram analysis method [Citation38–40].
2.2. Permeability coefficients
The permeability coefficient of a compound is obtained as an integral of its resistivity over the membrane normal, see Equation (Equation3
(3)
(3) ). While CG simulations provide access to the potential of mean force
, the acceleration of the dynamics introduced by CG models [Citation41] makes the estimation of the inhomogenous diffusivity
a priori difficult. However, atomistic simulations of several small molecules in a DOPC membrane showed that
is largely insensitive to the chemical detail [Citation15]. Moreover,
only provides a logarithmic correction to the
of a compound, see Equation (Equation3
(3)
(3) ). In our calculations we thus relied on a unique diffusivity profile, and parametrised it as
(4)
(4) with
,
,
,
, which correctly captures the main features of its atomistic counterpart [Citation15].
In the case of acidic compounds, accounting for deprotonation requires to slightly modify Equation (Equation3(3)
(3) ) to include the flux of both neutral and charged species. The total resistivity reads [Citation15]
, where
and
are the resistivities of the neutral and charged forms of the compound, respectively. The corresponding potentials of mean force
and
must be offset by the acid/base free-energy difference in bulk water [Citation36]
(5)
(5) where we consider the case of neutral
. Assuming that the diffusivity does not depend on the protonation state of the compound, the combination of the two PMFs in the total resistivity
can be represented by an effective potential
for all z, see Figure (c), from which
.
indicate the shifted counterparts of the neutral and charged PMFs, respectively.
Permeability coefficients predicted by the CG model were validated in Ref. [Citation23] against atomistic simulations results as well as experimental measurements for several chemically different small molecules covering a wide range of p. In both cases we observed a mean absolute error well within one
unit, demonstrating the accuracy of the CG model in reproducing the permeation rate of a compound.
3. Results
We considered all possible CG compounds consisting of a single Martini bead (‘unimers’) as well as those generated by binding together all possible combinations of two beads (‘dimers’), representative of a subset of the chemical space of small organic molecules in the range 30–160 Da [Citation22]. We calculated the PMF for all neutral unimers and dimers, 119 in total. For each neutral compound, we then determined
for its deprotonated counterpart. At the CG level, deprotonating a chemical group amounts to replacing the original bead type with a negatively charged one. We always represent deprotonated fragments by means of a
type, following the standard Martini notation. In the case of unimers, this choice automatically determines the CG representation of the deprotonated form of a compound. In the case of dimers, we initially assumed that deprotonation always occurs in the chemical fragment represented by the bead of higher polarity, but relax this assumption along the discussion, vide infra. By combining
and
in the total resistivity
, we calculated the permeability coefficient of every compound as a function of the ap
every 0.2 ap
unit. The set of permeability data was then projected on the
plane.
To analyze how the resulting permeabilities are affected by a change in the two molecular descriptors, we separately discuss the case of neutral compounds/weak acids () and strong acids (
).
3.1. Neutral compounds/weak acids: revisiting the Meyer-Overton rule
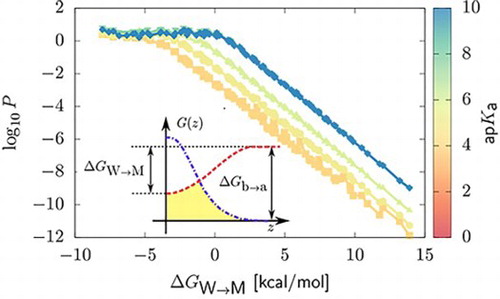
Figure displays a two-dimensional projection of the permeability data on the plane for neutral compounds and weak acids,
. We present results for a subset of ap
values, and colour the data accordingly. Panels (a) and (b) distinguish between the permeabilities obtained for Martini unimers and dimers, effectively amounting to a segregation in molecular weights [Citation22].
Figure 2. Two-dimensional projection of the permeability data on the plane in the case of neutral compounds and weak acids,
. Data are coloured according to the value of the ap
. Panels (a) and (b) distinguish between results obtained for Martini unimers and dimers. For neutral compounds with
, the permeability becomes independent on the value of the acid dissociation constant. Accordingly, we only show data up to
. We present permeability coefficients obtained from HTCG simulations (points) and the corresponding interpolated permeability profiles (lines). For neutral compounds, we include predictions from the Meyer-Overton rule in the presence of flat potentials of mean force with heights equal to
(‘Overton-mem’, dashed lines), see text.
Due to the finite number of interaction types in the Martini model, simulation results are located at a discrete set of partitioning free energies. In both cases, the regularity of the points at fixed ap as a function of
allows to reconstruct continuous permeability profiles. We include these interpolations in the corresponding panels of Figure . The independence of the interpolated curves on the CG representation, together with the dense covering in
generated by CG dimers, highlight the smoothness of the permeability coefficient as a function of ap
and
. These results enable the calculation of the permeation rate only starting from two thermodynamic properties, providing a phenomenological alternative to the Meyer-Overton rule in the framework of the accurate ISDM.
The permeability coefficient weights the contributions of both the charged and neutral
of a compound in an effective potential
which approximately dictates the shape of the total resistivity,
(Figure (c)). Understanding the smooth dependence of the permeability on both descriptors thus requires to more carefully analyze how the underlying potentials of mean force combine in
. For this purpose, within the class of weak acids we separately discuss the case of neutral compounds – or ‘extremely weak’ acids – with
, and weak acids,
.
3.1.1 Neutral compounds: 
For neutral compounds, the permeability is independent of the acid dissociation constant. This stems from the fact that by increasing the ap beyond
the charged repulsive PMF
is shifted towards increasingly higher values, and does not contribute to the effective PMF (Figure (c)). One obtains
, such that
.
For , Figure shows that
roughly plateaus. The reason is straightforward: apolar compounds are characterised by a negative, attractive well of
within the membrane core (Figure (b)). This well gives a negligible contribution to the integral in Equation (Equation3
(3)
(3) ) compared to the regions in which the PMF reaches saturation – i.e. as the compound approaches the bulk water environment.
Moving towards the repulsive nature of
(Figure (b)) increases the resistivity
, resulting in a reduction of the permeation rate. In this case, Figure highlights a linear dependence of
as a function of
. It was recently observed that for polar compounds the permeability is mainly dictated by the height of the repulsive PMF (i.e.
) irrespective of its detailed shape – e.g. width, presence of attractive tails [Citation16]. This suggests the linearity in Figure to be a consequence of this insensitivity.
We thus approximated the membrane as a bulk homogeneous solution, and employed the Meyer-Overton rule to calculate the corresponding permeabilities. In Equation (Equation2(2)
(2) ), we assumed
and set
equal to the average thickness and diffusivity values. The use of
amounts to replacing an ensemble of repulsive PMFs smoothly approaching zero by square-well potentials with heights equal to
. We stress that
differs from the partitioning coefficient K in Equation (Equation2
(2)
(2) ), which is instead calculated accounting for the inhomogeneous nature of the membrane, vide infra.
Permeabilities obtained from this homogenous model are included in both panels of Figure (dashed lines). In the case of polar compounds results only slightly underestimate ISDM ones, the maximum deviation being roughly one unit. This confirms that for
the permeability mainly depends on the height of the repulsive PMF [Citation16], and explains the linearity of
as a function of
. On the contrary, for
the homogeneous model strongly overestimates the permeability coefficient: this stems from the fact that the square-well potentials neglect the saturation of the PMFs that occur by approaching the bulk water environment – the only region that for apolar compounds contributes to the resistivity.
This homogeneous model differs from the Meyer-Overton rule, in which the partitioning coefficient K of a compound reads
(6)
(6) Predictions from the rule were recently shown to overestimate ISDM ones [Citation19], and the permeability coefficient was observed to be multivalued in K. By approximating
in Equation (Equation3
(3)
(3) ) with its average value, a comparison with Equation (Equation2
(2)
(2) ) highlights that in the latter we are replacing the inverse of the integral of
with the integral of its inverse. The regions contributing to the two integrals are essentially swapped: for apolar compounds the negative well becomes the major contributor to Equation (Equation6
(6)
(6) ), while in the ISDM its contribution is negligible compared to the region of saturation of the PMF. The opposite occurs in the case of polar compounds, where the free-energy barrier
that dictates permeation rate in the ISDM does now not contribute to Equation (Equation6
(6)
(6) ), which is instead governed by the regions where the PMF reaches saturation or exhibits attractive tails [Citation19]. This lies at the origin of the failure of the Meyer-Overton rule in reproducing ISDM permeabilities for neutral compounds.
3.1.2 Weak acids:
At fixed , Figure shows that weak acids with
are characterised by a reduction in the permeability coefficient with respect to neutral compounds. In addition to polarity, the deprotonation of a small molecule affects its permeation rate.
By decreasing the ap, both the neutral and charged PMFs of a compound contribute to the total resistivity. The neutral PMF
is shifted towards increasingly higher values by a factor of
, and the minimum between the two profiles dictates the shape of
(Figure (c) and Section 2.2), with
. We performed this construction for all compounds investigated in this work, resulting in the combination of a large variety of PMFs. However, Figure highlights that at fixed ap
the only differences between the permeabilities of acidic and neutral compounds as a function of
consist in the length of the initial plateau and in the presence of a negative offset for the linear regime.
To understand the origin of this behaviour, Figure displays two examples of the construction of for a polar and an apolar compound with
. For
(main plot in Figure ), combining the neutral and charged PMFs results in a repulsive
with an effective barrier given by the sum of
and
. As for polar compounds the permeability coefficient is mainly dictated by the height of the PMF, increasing this by a factor
– which is constant at fixed ap
and independent of
, see Figure (c) – preserves the linearity of
in
but generates the offsets observed in Figure .
Figure 3. Construction of (blue) for an acidic compound from its charged
(red) and neutral
(orange) PMFs. In calculating
,
is vertically shifted by
, and we consider
. We present results for a polar (main plot) and an apolar (inset) dimer.
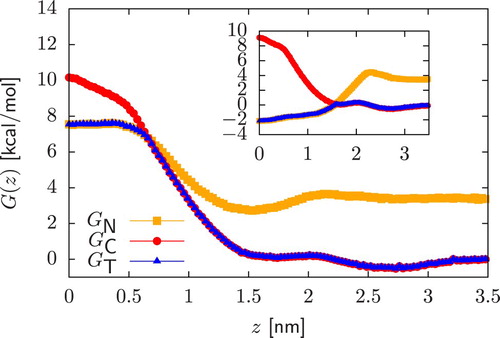
For (inset in Figure ), the effect of a small vertical shift of
for a fixed ap
is to reduce the attractive well that in the neutral case was not contributing to the resistivity. By increasing
, the well crosses zero and
becomes repulsive. At this stage, the permeability starts following the linear regime. This explains the reduction in the length of the plateau observed in the permeability profiles for
by increasing the strength of the acid.
Overall, our results suggest that in the case of weak acids the permeability is mainly dictated by the combination of two free-energy differences: the water/membrane partitioning free energy , and the acid/base free-energy difference
, which directly links to the ap
(Figure (c)). This is observed irrespective of the detailed shape of the underlying PMFs, and explains the smooth dependence of the permeability observed as a function of the two molecular descriptors.
The construction of allows for a further discussion about the role of the charged PMF of a compound in the permeation rate. Figure shows that if
has reached the shifted neutral PMF within the membrane core – a saturation condition always satisfied for
– an increase in its amplitude does not affect
. It follows that the permeability becomes largely independent on the polarity of the deprotonated form of the compound.
For a small molecule mapping to a CG dimer we originally assumed that deprotonation always occurred in the chemical fragment represented by the bead of higher polarity. We thus recalculated permeability coefficients relying on the opposite assumption – i.e. deprotonating the less polar bead. The resulting charged PMFs are characterised by a stronger repulsion within the membrane core, an example being shown in the inset of Figure .
Figure 4. Main plot: Permeability data of CG dimers ( scale) calculated by relying on different CG representations of the deprotonated form of a compound. Starting from a neutral dimer, coloured points are obtained by assuming that deprotonation always occur in the chemical fragment encapsulated in the bead of higher polarity. In dashed lines (‘Apolar-dep’) we always deprotonate the bead of lower polarity. Inset: Comparison of the charged PMFs
arising from the two different CG representations of the deprotonated form of the neutral polar dimer in Figure . The PMF of the compound obtained by deprotonating the bead of lower polarity is characterised by a stronger repulsion.
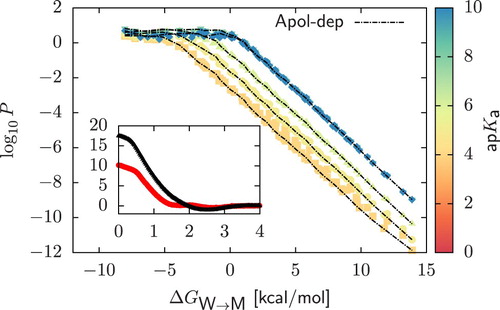
Permeability coefficients obtained from the two deprotonation prescriptions are presented in Figure (main plot). Results are essentially indistinguishable. Slight deviations are observed for , in which the condition of saturation of
is not always satisfied as we are moving towards strong acids – vide infra. Importantly, this analysis shows that while the permeation rate could in principle depend on which chemical fragment of the small molecule is subject to deprotonation, this does not matter in practice.
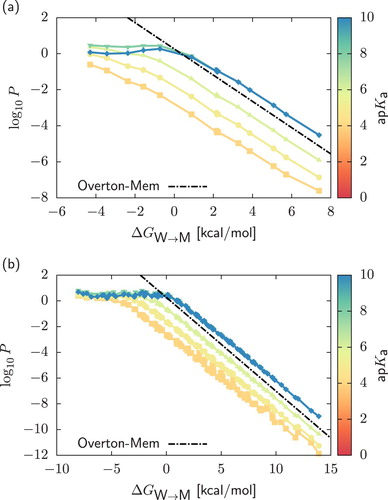
3.2. Strong acids: the role of amphiphilicity
Panel (a) of Figure displays permeability data of CG dimers as a function of for
, together with some previously analyzed results for
. At
, data initially follow a linear trend analogous to the one of weak acids due to the saturation of
in the effective PMF. However, for
we observe a spread of the points over the
plane: the permeability coefficient at fixed ap
becomes multivalued in
. This does not occur in the case of CG unimers, whose
continue to behave linearly as
increases (data not shown).
Figure 5. Panel (a): Two-dimensional projection of the permeability data on the plane in the case of strong acids with
. We include as reference a subset of the data obtained for weak acids,
. Points were obtained by assuming that starting from a neutral dimer, deprotonation always occur in the chemical fragment encapsulated in the bead of higher polarity. On the contrary, in dashed lines (‘Apolar-dep’) we always deprotonate the bead of lower polarity. Panel (b): Comparison of the neutral (main plot) and charged (inset) PMFs
and
of a
and a
dimers. In both cases, charged PMFs are obtained by assuming that deprotonation always occurs in the bead of higher polarity.
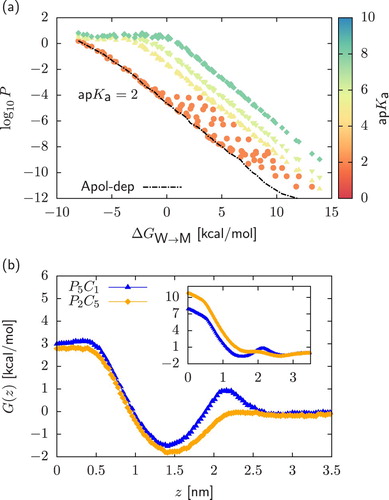
In strong acids, is such that neutral PMFs are shifted towards high values with respect to their charged counterparts (Figure (c)): only
contributes to the effective PMF, and
. Different permeabilities for a given
thus arise from compounds presenting very different polarities in their depronotonated forms, despite having similar ones in the neutral case – i.e. similar
.
Martini assumes additivity in the partioning free energy of its constituting beads. Many possible combinations of two neutral beads can result in the same : while these will lead to roughly the same free-energy difference for the neutral PMFs
, this does not necessarily hold for their deprotonated counterparts. As an example we consider a
dimer, composed by the most polar (P) and most apolar (C) Martini bead types, and a
dimer, in which we reduce the asymmetry in polarity between the two beads. Due to additivity, the compounds are characterised by similar neutral PMFs
(main plot in Figure (b)). Assuming that deprotonation always occurs in the bead of higher polarity, however, the charged form of the first compound shows a weaker repulsion from the membrane core (inset of Figure (b)). As
,
is characterised by a higher
:
compared to
for
.
The sensitivity of the permeability coefficient on the asymmetry in polarity between the chemical fragments composing the molecule suggests to introduce a third molecular descriptor. The most natural choice is the difference between the partitioning free energies of the two beads, , where beads are arranged in order of decreasing polarity. For a given
, increasing
amounts to decreasing/increasing the polarity of the more apolar/polar chemical group. As such,
is essentially quantifying the amphiphilicity of the small molecule.
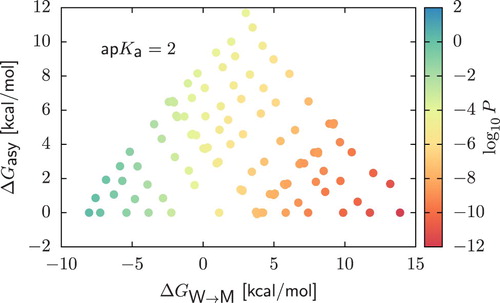
Permeability data for as a function of
and
are presented in Figure . The introduction of
completely solves the degeneracy in
observed in Figure (a): for a given
of the neutral form of the acid, increasing
generates an increase in the permeation rate. On the one hand, this is not surprising, meaning that the reduction in permeability generated by the deprotonation of a functional group can be mitigated by coupling it with a highly apolar one. However, in analyzing these results one has to account for the tendency of Martini to underestimate the PMF of charged compounds [Citation27,Citation42]. We thus repeated our calculations by assuming that deprotonation always occur in the bead of lower polarity, which as discussed in Section 3.1 generates an increase in the repulsive barrier of
. Results are included in Figure (a) (dashed line). In this case, the permeability coefficient follows a linear regime analogous to the case of weak acids due to saturation of the charged PMFs in
. We conclude that if the permeability coefficient depends on the amphiphilicity of the small molecule, due to the underestimation of the PMF of charged compounds in Martini this will be observed at extremely low values of the ap
. For strong acids, however, our results provide a lower bound for the permeation rate: at a fixed ap
, the permeability coefficient cannot be lower than the linear relation dictated by combining two free-energy differences,
and
.
Figure 6. Permeability coefficients ( scale) for CG dimers in the case of strong acids with
. We display results as a function of
and the asymmetry
.
is calculated as the difference between the water/membrane partitioning free energy of the beads composing the dimer, sorting them in order of decreasing polarity. In all cases, the charged form of the compound was obtained by assuming that deprotonation always occur in the bead of higher polarity.
4. Conclusions
While the Meyer-Overton rule links the permeability of a compound across a lipid bilayer to the partitioning coefficient, its validity is limited by the homogeneity assumption. An analogous relation in the framework of the accurate inhomogeneous solubility-diffusion model (ISDM) was missing up to now. In the ISDM, the permeability coefficient is a functional of the potential of mean force (PMF). Identifying this relation would require to probe large regions of chemical space to systematically investigate the impact of the PMF on the permeation rate. This is hampered by the overwhelming number of possible compounds.
In this work, we rely on CG simulations to systematically explore the permeability variations in a subset of the chemical space of small organic molecules, focussing on acidic compounds. We connect permeability coefficients calculated from the ISDM to two thermodynamic properties: the water/membrane free energy, , and acid dissociation constant, ap
. Results are obtained by probing a large variety of small molecules with very different PMFs, both neutral and charged. We carefully analyze how – and when – combining together these profiles can result in a smooth dependence of the permeability on the two thermodynamic variables.
For neutral compounds, the permeation rate is independent on the ap and smoothly depends on
. This is due to the fact that the permeability coefficient is mainly dictated by
irrespective of the detailed shape of the neutral PMF. The deviation of the Meyer-Overton rule from ISDM predictions stems from incorrectly accounting for this free-energy difference. For weak acids, the deprotonation of a chemical group affects the permeation rate of a compound. In this case, charged and neutral PMFs combine in an effective profile whose water/membrane free-energy difference is the sum of
and the acid/base free-energy difference
, the latter being a linear function of the ap
. The insensitivity of the permeability coefficient on the detailed shape of the effective PMF results in a smooth dependence of the permeation rate on our thermodynamic variables. Interestingly, we show that in weak acids the permeability is independent of the chemical fragment subject to deprotonation. For strong acids, the permeability is no longer a smooth function of the two original molecular descriptors. This requires to introduce a third orthogonal variable characterising the amphiphilicity of a small molecule. In this case, our results provide a lower bound to the permeation rate of a compound: the permeability cannot be smaller than a linear relation generated by the combination of
and
.
Acknowledgments
The authors thank Alessia Centi and Omar Valsson for critical reading of the manuscript and useful comments.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Roberto Menichetti http://orcid.org/0000-0002-5961-6438
Tristan Bereau http://orcid.org/0000-0001-9945-1271
Additional information
Funding
References
- I. Kola and J. Landis, Nat. Rev. Drug Discov. 3 (8), 711 (2004). doi: 10.1038/nrd1470
- W. Stein, Transport and Diffusion Across Cell Membranes (Elsevier, Amsterdam, 2012).
- K. Sugano, M. Kansy, P. Artursson, A. Avdeef, S. Bendels, L. Di, G.F. Ecker, B. Faller, H. Fischer, G. Gerebtzoff, H. Lennernaes and F. Senner, Nat. Rev. Drug Discov. 9 (8), 597 (2010). doi: 10.1038/nrd3187
- E. Awoonor-Williams and C.N. Rowley, Biochim. Biophys. Acta Biomembr. 1858 (7), 1672–1687 (2016). doi: 10.1016/j.bbamem.2015.12.014
- C. Pidgeon, S. Ong, H. Liu, X. Qiu, M. Pidgeon, A.H. Dantzig, J. Munroe, W.J. Hornback and J.S. Kasher, J. Med. Chem. 38 (4), 590–594 (1995). doi: 10.1021/jm00004a004
- M. Yazdanian, S.L. Glynn, J.L. Wright and A. Hawi, Pharm. Res. 15 (9), 1490–1494 (1998). doi: 10.1023/A:1011930411574
- M. Kansy, F. Senner and K. Gubernator, J. Med. Chem. 41 (7), 1007–1010 (1998). doi: 10.1021/jm970530e
- H. Meyer, Archiv für experimentelle Pathologie und Pharmakologie 42 (2–4), 109–118 (1899). doi: 10.1007/BF01834479
- C.E. Overton, Studien über die Narkose zugleich ein Beitrag zur allgemeinen Pharmakologie (Fischer, Jena, 1901).
- A. Missner and P. Pohl, ChemPhysChem 10 (9–10), 1405–1414 (2009). doi: 10.1002/cphc.200900270
- J.M. Diamond and Y. Katz, J. Membrane Biol. 17 (1), 121–154 (1974). doi: 10.1007/BF01870176
- S.J. Marrink and H.J. Berendsen, J. Phys. Chem. 98 (15), 4155–4168 (1994). doi: 10.1021/j100066a040
- L.W. Votapka, C.T. Lee and R.E. Amaro, J. Phys. Chem. B 120 (33), 8606–8616 (2016). doi: 10.1021/acs.jpcb.6b02814
- G. Parisio, M. Stocchero and A. Ferrarini, J. Chem. Theory Comput. 9 (12), 5236–5246 (2013). doi: 10.1021/ct400690t
- T.S. Carpenter, D.A. Kirshner, E.Y. Lau, S.E. Wong, J.P. Nilmeier and F.C. Lightstone, Biophys. J.107 (3), 630–641 (2014). doi: 10.1016/j.bpj.2014.06.024
- C.T. Lee, J. Comer, C. Herndon, N. Leung, A. Pavlova, R.V. Swift, C. Tung, C.N. Rowley, R.E. Amaro, C. Chipot, Y. Wang and J.C. Gumbard, J. Chem. Inf. Model. 56 (4), 721–733 (2016). doi: 10.1021/acs.jcim.6b00022
- O. De Vos, R.M. Venable, T. Van Hecke, G. Hummer, R.W. Pastor and A. Ghysels, J. Chem. Theory Comput. 14 (7), 3811–3824 (2018). doi: 10.1021/acs.jctc.8b00115
- R. Sun, Y. Han, J.M. Swanson, J.S. Tan, J.P. Rose and G.A. Voth, J. Chem. Phys. 149 (7), 072310 (2018). doi: 10.1063/1.5027004
- D.J. Smith, G. Leal, S. Mitragotri and M.S. Shell, J. Phys. D 51, 294004 (2018). doi: 10.1088/1361-6463/aacac9
- B.J. Bennion, N.A. Be, M.W. McNerney, V. Lao, E.M. Carlson, C.A. Valdez, M.A. Malfatti, H.A. Enright, T.H. Nguyen, F.C. Lightstone and T.S. Carpenter, J. Phys. Chem. B 121 (20), 5228–5237 (2017). doi: 10.1021/acs.jpcb.7b02914
- C.M. Dobson, Nature 432 (7019), 824–828 (2004). doi: 10.1038/nature03192
- R. Menichetti, K.H. Kanekal, K. Kremer and T. Bereau, J. Chem. Phys. 147 (12), 125101 (2017). doi: 10.1063/1.4987012
- R. Menichetti, K.H. Kanekal and T. Bereau, ACS Cent. Sci. 5 (2), 290–298 (2019). doi: 10.1021/acscentsci.8b00718
- W. Noid, J. Chem. Phys. 139 (9), 09B201_1 (2013). doi: 10.1063/1.4818908
- S.J. Marrink, A.H. de Vries and A.E. Mark, J. Phys. Chem. B 108, 750–760 (2004). doi: 10.1021/jp036508g
- S.J. Marrink, H.J. Risselada, S. Yefimov, D.P. Tieleman and A.H. de Vries, J. Phys. Chem. B 111, 7812–7824 (2007). doi: 10.1021/jp071097f
- L. Monticelli, S.K. Kandasamy, X. Periole, R.G. Larson, D.P. Tieleman and S.J. Marrink, J. Chem. Theory Comput. 4, 819–834 (2008). doi: 10.1021/ct700324x
- T. Bereau and K. Kremer, J. Chem. Theory Comput. 11 (6), 2783–2791 (2015). doi: 10.1021/acs.jctc.5b00056
- R. Menichetti, K. Kremer and T. Bereau, Biochem. Biophys. Res. Commun. 498 (2), 282–287 (2018). doi: 10.1016/j.bbrc.2017.08.095
- S.J. Marrink and D.P. Tieleman, Chem. Soc. Rev. 42 (16), 6801–6822 (2013). doi: 10.1039/c3cs60093a
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput. 4, 435–447 (2008). doi: 10.1021/ct700301q
- M. Parrinello and A. Rahman, J. Appl. Phys. 52 (12), 7182–7190 (1981). doi: 10.1063/1.328693
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys. 126 (1), 014101 (2007). doi: 10.1063/1.2408420
- T.A. Wassenaar, H.I. Ingølfsson, R.A. Böckmann, D.P. Tieleman and S.J. Marrink, J. Chem. Theory Comput. 11 (5), 2144–2155 (2015). doi: 10.1021/acs.jctc.5b00209
- G.M. Torrie and J.P. Valleau, J. Comput. Phys. 23 (2), 187–199 (1977). doi: 10.1016/0021-9991(77)90121-8
- J.L. MacCallum, W.D. Bennett and D.P. Tieleman, Biophys. J. 94 (9), 3393–3404 (2008). doi: 10.1529/biophysj.107.112805
- T. Bereau, Z.J. Wang and M. Deserno, J. Chem. Phys. 140 (11), 115101 (2014). doi: 10.1063/1.4867465
- S. Kumar, J.M. Rosenberg, D. Bouzida, R.H. Swendsen and P.A. Kollman, J. Comput. Chem. 13 (8), 1011–1021 (1992). doi: 10.1002/jcc.540130812
- T. Bereau and R.H. Swendsen, J. Comput. Phys. 228 (17), 6119–6129 (2009). doi: 10.1016/j.jcp.2009.05.011
- J.S. Hub, B.L. De Groot and D. Van Der Spoel, J. Chem. Theory Comput. 6 (12), 3713–3720 (2010). doi: 10.1021/ct100494z
- J.F. Rudzinski, K. Kremer and T. Bereau, J. Chem. Phys. 144 (5), 051102 (2016). doi: 10.1063/1.4941455
- S.O. Yesylevskyy, L.V. Schäfer, D. Sengupta and S.J. Marrink, PLOS Comput. Biol. 6 (6), e1000810 (2010). doi: 10.1371/journal.pcbi.1000810