

Abstract
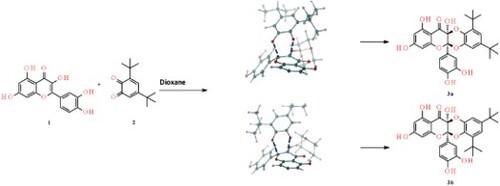
A theoretical mechanistic investigation of the cycloaddition reaction of quercetine with 3,5-di-tert-butyl-o-quinone has been carry out employing density functional calculations at M06-2X/6-31G(d,p) level of theory. This computational study was performed in order to gain insight in the energetic and structural parameters that rule the observed experimental selectivity in the synthetised cyclodimers obtained under classical heating and microwave irradiation conditions and the influence of the solvent on their chemical reactivity. The effect of the solvent in the selectivity was studied implicitly using the polarisable continuum model in absence and presence of one explicit solvent’s molecule. The theoretical results explain the main issues of the classical heating and the MWI reaction and suggest that thermal effects are directly responsible for the reaction selectivity observed. The comparison of the mechanistic pathway and the non-covalent interactions, shows that a dioxane assisted mechanism can explain the observed experimental selectivity of the two synthetised compounds, thus the dioxane molecule play a catalytic role on this hetero Diels–Alder reaction.
GRAPHICAL ABSTRACT
KEYWORDS:
Acknowledgments
The authors are grateful to Romina Quijano and Oliver Lown for the English review of the manuscript, and to Dirección General de Servicios de Cómputo Académico, Universidad Nacional Autónoma de México, for supercomputer time.
Disclosure statement
No potential conflict of interest was reported by the authors.