

Abstract
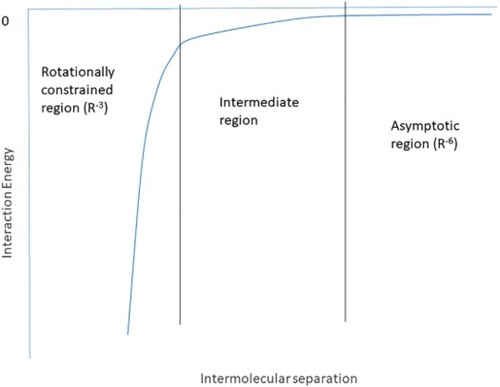
Dispersion is a ubiquitous intermolecular force that affects short- and long-range potentials between molecules. Dispersion affects crystallisation, self-assembly, enzyme selectivity, and surface interactions, among countless other processes. Hence, it is of interest to be able to quantify dispersion when considering intermolecular potentials using quantum chemical techniques. This communication presents a first-principles method for computing the dispersion interaction quantum chemically. London’s description of dispersion is strictly only valid at large intermolecular separations since it is predicated on the free mutual rotation of molecules. We recast the description of London’s dispersion interaction in modern terminology and show that dispersion is caused by the correlation of electrons on different molecules and is hence attractive at all intermolecular separations. The discussion below extends the description of dispersion to all separations and orientations. This permits computation of instantaneous dispersion in systems with constrained mutual rotation and the formation of dispersion aggregates. We also show that ab initio correlated methods include dispersion effects naturally and discuss the balanced treatment of dispersion using these techniques. Finally, we propose a natural extension to the method to include quadrupolar and octupolar dispersion interactions.
GRAPHICAL ABSTRACT
Acknowledgment
Computational resources for this study were made available by ACENET (www.ace-net.ca) and Compute Canada (www.computecanada.ca).
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes
1 Second-order Møller-Plesset terms have the form .