

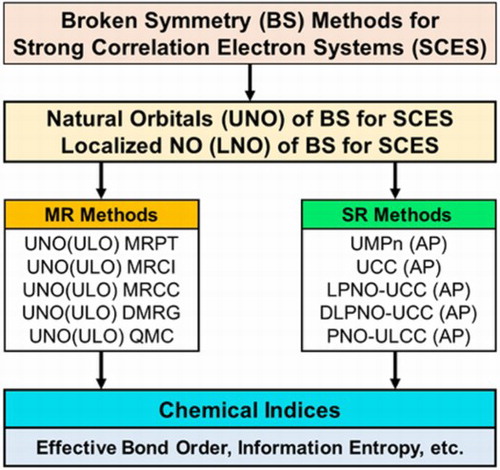
ABSTRACT
Domain-based local pair natural orbital (DLPNO) coupled cluster single and double (CCSD) methods with perturbative triples (T) correction with NormalPNO were used to compute energies for twelve different S1 structures of the CaMn4O5 cluster in the oxygen evolving complex (OEC) of photosystem II (PSII). The DLPNO-CCSD(T0) calculations with TightPNO for the important six structures among them revealed that the right (R)-opened S1XYZW structures were more stable than the corresponding left (L)-opened structures (X = O(5), Y = W2, Z = W1, and W = O(4)) of CaMn4O5. The three different S1 structures belonging to the R-opened type (S1acca, S1bbca, and S1abcb, where O2- = a, OH- = b and H2O = c) were found nearly degenerated in energy, indicating the possibility of the coexistence of different structures in the S1 state. The DLPNO-CCSD(T0) calculations with TightPNO supported the proposal of a dynamic equilibrium model based on the multi-intermediate structures for the S1 state, which is also in agreement with EPR and other experimental and hybrid DFT computational results. Implications of the computational results are discussed in relation to scope and applicability of NormalPNO and TightPNO for the CCSD(T0) calculations of strongly correlated electron systems such as 3d transition-metal complexes.
GRAPHICAL ABSTRACT
Acknowledgements
This work has been supported by MEXT KAKENHI Grant Nos. JP18H05154, JP18H05167, and JP17H06434 (MS, HI, KY) and by FOCUS Establishing Supercomputing Center of Excellence. Numerical calculations were carried out with the support of the Research Center for Computational Science, Okazaki, Japan.
Disclosure statement
No potential conflict of interest was reported by the authors.