
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Systems composed of soft matter (e.g. liquids, polymers, foams, gels, colloids, and most biological materials) are ubiquitous in science and engineering, but molecular simulations of such systems pose particular computational challenges, requiring time and/or ensemble-averaged data to be collected over long simulation trajectories for property evaluation. Performing a molecular simulation of a soft matter system involves multiple steps, which have traditionally been performed by researchers in a ‘bespoke’ fashion, resulting in many published soft matter simulations not being reproducible based on the information provided in the publications. To address the issue of reproducibility and to provide tools for computational screening, we have been developing the open-source Molecular Simulation and Design Framework (MoSDeF) software suite. In this paper, we propose a set of principles to create Transparent, Reproducible, Usable by others, and Extensible (TRUE) molecular simulations. MoSDeF facilitates the publication and dissemination of TRUE simulations by automating many of the critical steps in molecular simulation, thus enhancing their reproducibilitya. We provide several examples of TRUE molecular simulations: All of the steps involved in creating, running and extracting properties from the simulations are distributed on open-source platforms (within MoSDeF and on GitHub), thus meeting the definition of TRUE simulations.
GRAPHICAL ABSTRACT

1. Introduction
Reproducibility in scientific research has become a prominent issue, to the extent that some have opined that science has a ‘reproducibility crisis’ [Citation1]. Along with the rest of the scientific community, computational scientists are grappling with the central question: How can a computational study be performed and published in such a way that it can be replicated by others? This has become increasingly important as researchers seek to harness the ever expanding computational power to perform large-scale computational screening of materials [Citation2–8] inspired by the materials genome initiative (MGI) [Citation9], where reproducibility issues commonly faced in small-scale studies will only be compounded as the number of simulations grow by orders of magnitude.
Addressing the issues of reproducibility in soft matter simulation is particularly challenging, given the complexity of the simulation inputs and workflows. Here, we define soft matter as anything easily deformed at room temperature, e.g. liquids, polymers, foams, gels, colloids, and most biological materials. Figure shows a schematic of the general multi-step workflow for performing atomistic simulations of soft matter systems, proceeding from system ‘chemistry’ (chemical composition and state conditions such as phases(s), temperature, pressure, and composition) to ‘properties’ (e.g. structural, thermodynamic and transport properties, phase equilibria, and dielectric properties). In such systems, the differences in potential energy between distant configurations are on the same order as the thermal motion, requiring time and/or ensemble-averaged data to be collected over long simulation trajectories for property evaluation. The equilibration procedures and system sizes considered may strongly influence the resulting measured properties, since one must consider both the local conformations of the underlying components, along with any mesoscopic structuring present in the system. To capture sufficiently large length and time scales, soft matter simulations are typically performed using methods such as molecular dynamics (MD) or Monte Carlo (MC) that employ empirical force fields to model the interactions between atoms and molecules; the appropriate force field parameters must be identified before the simulation can be performed.
Figure 1. Schematic of the typical process required to compute properties of soft matter systems from system ‘chemistry’, which refers to chemical composition and state (including temperature, pressure and composition), starting from the need to either gather or derive force field parameters to model the system. For coarse-grained (CG) simulations, the CG force fields are often derived from atomistic simulations.
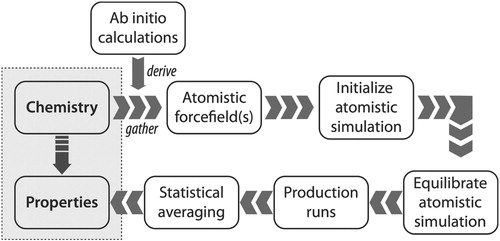
Some commonly available force fields, such as the Optimized Potentials for Liquid System (OPLS) [Citation10] and the General Amber Force Field (GAFF) [Citation11] contain thousands of possible parameters that are differentiated by their chemical context (e.g. the element a given interaction site represents, the number and identity of bonded neighbours, the local environment of bonded neighbours, the type of system, etc.). Selecting the appropriate force field parameters for a particular use case is often non-trivial. Workflows may also involve the optimisation of specific parameters, such as partial charges, or require separate procedures to develop novel force fields, such as coarse-grained (CG) models, before a simulation can be performed. Furthermore, due to the complex nature of the underlying constituents (e.g. highly branched polymers), setup of an initial system configuration may be challenging and require additional relaxation procedures to ensure system stability [Citation12].
As such, soft matter simulations typically require multi-step workflows with many inputs. The various steps are often accomplished by separate pieces of syntactically and/or semantically incompatible software tools that may require translators or ad hoc modifications to facilitate interoperability. These tools, and especially the ‘glue’ code that facilitates interoperability, are typically neither publicly available nor version-controlled. The latter is particularly important for long-term reproducibility, since to repeat a particular calculation may require using versions of the relevant codes used when the work was originally published, which could be a number of years ago.
The above complexities often make it difficult for researchers themselves to fully capture and preserve the procedures used to perform a simulation, let alone clearly disseminate these to the rest of the community. A typical soft matter simulation publication provides an overview of the methods and procedures used but falls significantly short of including the necessary information to unambiguously reproduce the published work. This information includes, but is not limited to, citations to the sources of force field(s) used, the numeric parameters of the force field(s) used, how the force field parameters were assigned to the system, constants and options provided in the underlying algorithms, and the exact choices used in constructing the initial configuration of the system. It is important to recognise that the results from a simulation can depend on the minute details [Citation13]. These details include, but are not limited to, the random seed used to generate a distribution, the specific force field parameters and how they were used, the exact procedures employed to equilibrate a system, etc. For example, small variations in force fields (e.g. changes in distances at which interactions are truncated, different partial charges, the specific method for handling long-ranged interactions, etc.) can change some predicted properties quite significantly [Citation13–15]. The minute details may also be inherent to the software used to perform the simulations, and thus the use of ‘in-house’ or commercial (i.e. closed-source) software stymies reproducibility. If the source code cannot be viewed, the underlying algorithms and inputs cannot be examined, the quality of the code and whether it has undergone proper validation is unknown, and errors cannot be identified. As an example, a long-standing disagreement related to phase transitions in supercooled water was only recently settled after the source code of the in-house software used to perform the calculations was shared. The differences in observed transitions were attributed to a subtle error in how velocities were assigned when initialising the many short MD simulations in the hybrid MD/MC workflows [Citation16,Citation17]. The use of open-source simulation engines therefore clearly enhances reproducibility, as the underlying source code can be examined (note, the use of open-source simulation engines is now routine for MD studies, but often these engines and other open-source codes are modified to implement new force field parameters or functional forms, and MC studies still commonly use in-house software). However, it is atypical for input scripts and data files for open-source simulation engines to be included as part of a publication and thus reproducibility still largely depends on the thoroughness of the description of the methods and model in the text. Furthermore, the algorithms and specific choices used to generate a data file, which may influence the results and their validity (e.g. how a force field was applied), are lost if the software and/or procedures used to generate the data file are not made available. Even when using open-source simulation engines, researchers still routinely use in-house software for other steps in the process, i.e. generation of initial configurations, selection of force field parameters, and analysis. Furthermore, if a workflow relies upon manipulation or modification of individual pieces of software by a user (e.g. initialising a system using software with a graphical user interface, GUI [Citation18]), or human-modification of files, it is often difficult to capture and convey the exact procedures in such a manner that they can be reproduced by another researcher.
Fortuitously, several researchers have proposed general guidelines for increasing reproducibility in computational research, which can be used to infer best practices for soft matter simulation. Donoho et al. [Citation19] propose that all details of computations – code and data – should be made ‘conveniently available’ to other researchers; they also provide arguments in favour of the creation and use of community developed software libraries and the use of scripting. Others [Citation20,Citation21] have proposed succinct ‘rules’ as keys to reproducible computational research, including version control, replacement of manual input with scripts, and public access to these scripts, input files, and resulting data. It was also noted that computational frameworks that integrate different tools within a common environment naturally satisfy many of these rules. One of the most vocal proponents of reproducibility in computational science [Citation22,Citation23] has gone as far as asserting that GUIs are the ‘enemy of reproducibility’. GUIs hide details and require human interaction and manipulation in contrast to scripts, which fully reveal the way in which calculations are performed. A classic example is Excel spreadsheets, where the relationship between calculation cells and data is normally hidden, and the order of calculation is not obvious, nor necessarily controllable. In 2010, Harvard University economists Reinhart and Rogoff published a highly cited and influential paper on the role of debt in limiting growth in national economies [Citation24]. The study, based on data manipulated within an Excel spreadsheet, was often cited by politicians favouring austerity policies in the wake of the 2008 financial crisis while public economic policy was being formed. Subsequently, Herndon et al. [Citation25] found that the spreadsheet contained errors in formulae that dramatically changed the conclusions.
Determining how these guidelines for reproducibility should be – and/or can be – implemented in soft matter simulation is in itself a challenge. For example, simply providing code is not effective if that code is poorly written or not well documented and has subtle issues, such as dependencies within a code (e.g. use of external libraries, especially if they are proprietary/non-free or difficult to obtain/install). These issues may create barriers to proper compilation/installation and hence hamper reproducibility. Similarly, providing a raw data file without defining the structure of it, and/or without appropriate metadata, does little to aid in reproducibility. Since journals largely do not provide mechanisms for sharing code, scripts, and/or data (aside from supplemental material), it is also not clear how such information should best be shared.
As such, in order to implement best practices, we assert that the development of new tools and standards will be required, in order to facilitate necessary changes to the way in which simulators perform and publish their research. However, development of new tools does little to improve reproducibility if those tools are not used; to be widely adopted by the community, they must provide additional value to researchers, e.g. minimising errors, reducing development time, preventing knowledge loss, providing novel functionality, etc.
For almost a decade now, we have been developing a robust Python-based, open-source integrated software framework for performing simulations of soft matter systems with the goal of implementing best practices and enabling reproducibility. This framework, known as the Molecular Simulation and Design Framework (MoSDeF) [Citation26], was developed initially at Vanderbilt University, in collaboration with computer scientists in the Institute for Software Integrated Systems [Citation27], to facilitate screening studies of monolayer lubrication using MD methods. MoSDeF provides a core foundation and includes tools for programmatic system construction (mBuild) [Citation28,Citation29], tools for encoding force field usage rules and their application (Foyer) [Citation30–32], and has recently integrated the signac framework [Citation33,Citation34], developed at the University of Michigan as a means of improved data and workflow management. The MoSDeF toolkit has been used in various published results [Citation7,Citation8,Citation28,Citation32,Citation35,Citation36] and ongoing research projects, with the primary MoSDeF tools having each been downloaded over 18,000 times from Anaconda Cloud [Citation37] since February 2017. Despite being initially developed for monolayer lubrication, the underlying tools can be and have been applied to soft matter systems in general, and the modular, object-oriented design naturally allows for intuitive extension. Current MoSDeF activities are expanding the capabilities related to:
Initializing system configurations by providing a plugin architecture for community contributions
Providing initialisation routines for a wide variety of common systems
Developing an improved backend that will support an increased number of force field types and simulation engines, including open-source MC software
Developing modules that implement methods for partial charge assignment
Including improved support and libraries for coarse-grained models
Developing modules that allow for reproducible derivation of coarse-grained and atomistic force fields
Developing workflows for free energy methods and phase equilibria
Specifically identifying and implementing best practices within the various modules/workflows that improve reproducibility.
Through the MoSDeF integrated framework, the exact procedures used to set up and perform simulation workflows and associated metadata (i.e. the provenance) can be scripted, encapsulated, version-controlled, preserved, and later reproduced by other researchers. This allows molecular simulation studies to be conducted and published in a manner that is TRUE: Transparent, Reproducible, Usable by others, and Extensible.
The remainder of this paper is organised as follows. In Section 2, we briefly review MoSDeF an its capabilities. In Section 3, we consider four examples of TRUE molecular simulations in diverse application areas. Finally, in Section 4, we summarise our conclusions and prospects for future development of MoSDeF.
2. Overview of MoSDeF
2.1. MoSDeF tools and capabilities
MoSDeF is a set of an open-source Python libraries, designed to facilitate the construction and parameterisation of systems for molecular simulation. MoSDeF includes routines to output syntactically correct configuration files in formats used by common simulation engines, currently supporting GROMACS [Citation38–40], LAMMPS [Citation41], HOOMD-blue [Citation42,Citation43], and Cassandra [Citation44], as well as other common file formats (e.g. MOL2, PDB) through integration with the open-source ParmEd [Citation45] parameter editing package. Each library (i.e. Python module) in MoSDeF is designed such that it can be used as a standalone package, in combination with other libraries within MoSDeF, or with other libraries developed and used by the community. This composability/modularity is an essential design feature in terms of the robust development of MoSDeF, allowing the framework to be more modifiable, testable, extensible, and have fewer bugs than monolithic approaches [Citation13]. MoSDeF is implemented using concepts from the computer science/software engineering field of model integrated computing (MIC) [Citation46,Citation47], a systems engineering approach, pioneered at the Institute for Software Integrated Systems (ISIS) at Vanderbilt, that emphasises the creation of domain-specific modelling languages that capture the features of the individual components of a given process, at the appropriate level of abstraction. By using concepts from MIC, MoSDeF can easily be abstracted and is able to capture the relationships that exist between data and processes regardless of the level of abstraction, essential for ensuring that system initialisation scripts are transparent and usable by others. MoSDeF follows a modern open-source development model with special emphasis on effective code sharing, accepting external feedback, and bug reporting.
All modules and workflows developed for MoSDeF build upon the scientific Python stack, thus enabling transparency, promoting code reuse, lowering barriers to entry for new users, and promoting further community driven, open-source development.
GitHub is used for hosting MoSDeF's version-controlled software development, deployment, and documentation/tutorials, using a pull request (i.e. fork-pull) model that allows for code review and automated testing, helping ensure proper standards have been followed and allows for automated testing of software and software artifacts.
Automated builds and testing of the software are hosted on Travis CI [Citation48] and also on Microsoft's Azure Pipelines [Citation49] to ensure that proposed modifications to the code do not break the current performance and the CodeCov [Citation50] tool is used to ensure that modifications to the code are covered by unit tests.
All software developed as part of the MoSDeF project are open-source, with the standard MIT license [Citation51] that allows free use, reuse, modifications, as well as commercialisation.
Slack [Citation52] is used to facilitate effective collaborative communication and software development across a wide geographic area [Citation53].
By developing software in a modular, extensible, open-source manner, using freely available tools designed for collaborative code development (e.g. git, GitHub, and Slack), we are creating a long-term community-developed effort, similar to the success seen by other tools in the community (e.g. GROMACS [Citation38–40], VMD [Citation54], LAMMPS [Citation41], HOOMD-blue [Citation42,Citation43], etc.). This has become especially important as the group of MoSDeF developers has expanded beyond Vanderbilt University. A recent U.S. National Science Foundation grant [Citation55] has provided support for leading molecular simulation research groups from Vanderbilt, the universities of Michigan, Notre Dame, Delaware, Houston and Minnesota, along with Boise State University and Wayne State University to further improve and increase support of MoSDeF as described below. This group spans a broad range of expertise, and an equally broad range of scientific applications, open-source simulation codes (HOOMD-blue [Citation42,Citation43], Cassandra [Citation44], GOMC [Citation56,Citation57] and CP2K [Citation58]), workflow and data management software [Citation33,Citation34,Citation59] and algorithms and analysis tools; computer scientists from ISIS are also involved in the collaboration, helping to ensure the use of best practices and provide novel insight into algorithmic and software development. In combination, this collaboration is working to dramatically expand the capabilities of MoSDeF and thus facilitate researchers in the area of molecular simulation to be able to publish TRUE simulations.
Here, we briefly describe the two key tools used in the current version of MoSDeF, focussing on the specific aspects of the tools that help to enable TRUE simulations.
2.1.1. mBuild
The mBuild Python library [Citation28,Citation29] is a general purpose tool for constructing system configurations in a programmatic (i.e. scriptable) fashion. While tools exist in the community for system construction [Citation60–62], they tend to be system specific (e.g. bilayer construction), often employ GUIs which may hamper reproducibility [Citation22] and may be limiting for workflows that require automation, and most are designed around the concept that components of the system can be described by self-contained templates (e.g. where a system can be constructed by simply duplicating a template that describes a molecule). Such existing tools have typically not been designed to work for systems where bonds are added between different components (e.g. polymer grafted surfaces) or for systems where one component is semi-infinite (e.g. a silica substrate that is periodic in-plane) and most do not allow programmatic variation of specific structural/chemical aspects (e.g. the length of a polymer, the polymer repeat unit, size of a substrate, etc.); mBuild was designed specifically to provide this missing functionality.
Rather than providing a tool to perform initialisation that only applies to a specific family of systems (e.g. monolayers), mBuild provides a library of functions that users can combine, extend, and add to, in order to construct specific systems of interest. mBuild allows users to hierarchically construct complex systems from smaller, interchangeable pieces that can be connected, through the use of the concept of generative, or procedural, modelling [Citation28]. This is achieved through mBuild's underlying Compound data structure, which is a general purpose ‘container’ that can describe effectively anything within the system: an atom, a coarse-grained bead, a collection of atoms, a molecule, a collection of Compounds, or operations (e.g. a Compound that includes a routine to perform polymerisation). To join Compounds (e.g. attachment of a Compound that defines a polymer to a Compound that defines a surface), Compounds can include Ports that define both the location and orientation of a possible connection. In mBuild, a user (or algorithm) defines which Ports on two Compounds should be connected and the underlying routines in the software automatically performs the appropriate translations and orientations of the Compounds (see Klein et al. [Citation28] for more details). This creates a new (composite) Compound that contains both of the original Compounds, now appropriately oriented and positioned in space, with an explicit bond between them; since Compounds are general data structures, the same operations (rotation, translation, connecting of Ports, etc.) that were performed on the underlying Compounds can be performed on this new composite Compound. The mBuild library can be used to create systems from ‘scratch’ whereby a user implements all the relevant code to define the building blocks and how they should be connected, or by using and/or extending the various ‘recipes’ included in mBuild. mBuild includes (but is not limited to) ‘recipes’ for initialising polymers, tilings (e.g. duplicating a unit cell, including bonding information), patterning (disk, sphere, random, etc.), lattices either from a Crystallographic Information File (CIF), their Bravais lattice parameters, or the vectors describing the prism, box filling (via integration with PACKMOL [Citation63]), monolayers and brushes on flat, curved, and spherical surfaces, and bilayers and lamellar structures.
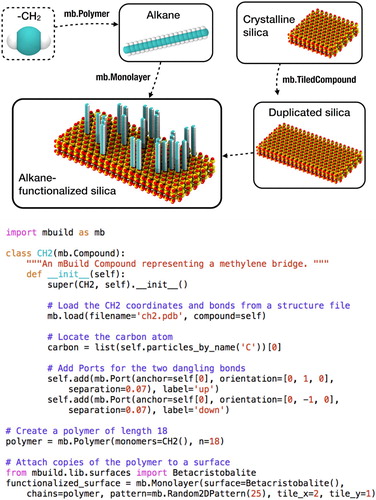
As an example, Figure shows a schematic and associated code for the construction of an alkane grafted silica surface. In this code, a custom Compound class is defined for a CH moiety alongside a Compound from the mBuild library that defines a crystalline silica surface; a ‘recipe’ included in mBuild that performs polymerisation (Polymer) is used to connect copies of the CH
moieties; the Monolayer ‘recipe’, also included in mBuild, is used to perform the functionalization of the silica surface with the polymer, returning a single composite Compound of the functionalised surface (for readability, the terminal groups are ignored in this example). This example also highlights how system construction can be programmatically varied, e.g. the Polymer class takes as input the number of repeat units (in this case, set to 18). Similarly, the size of the substrate can be toggled in the Monolayer class, where tile_x and tile_y define the number of times the substrate is duplicated in the respective dimension. The number of chains attached to the surface can also be modified via the number passed to the Random2DPattern class (here set to 25). Because Compounds are general containers, changes to, e.g. the length of the polymer, do not require changes to the rest of the script, namely the Monolayer class. Similarly, characteristics such as the repeat unit passed to the Polymer class can be readily changed without need to change other aspects of the script. As a result, by using the mBuild library, complex system initialisation and variation/extension can often be accomplished without the need to write significant amounts of code. As this example shows, by using concepts from MIC, construction of systems in mBuild can be readily abstracted (i.e. the level of complexity reduced) to the level most appropriate to describe (i.e. model) the system, without making system construction a ‘black box’. Since mBuild is an open-source, freely available Python library, scripts that unambiguously define all the steps needed to initialise a system can be easily shared and disseminated with publications, with all code easily interrogated, allowing system construction to be reproduced and improving transparency; mBuild has additionally been architected so that users can contribute custom ‘recipes’ for system initialisation via a plug-in environment, further allowing such routines to be easily shared, utilised and extended by others.
Figure 2. Python script that uses mBuild to define a class for a –CH– group, create a polymer composed of multiple –CH
– groups, and connects copies of this polymer to a surface. Note for simplicity, the terminal CH
group is not shown.
2.1.2. Foyer
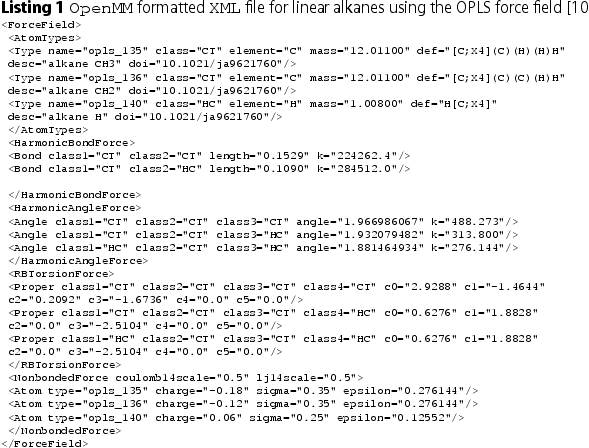
The Foyer library [Citation30] is a tool for applying force fields to molecular systems (i.e. atom-typing). Foyer provides a standardised approach to defining chemical context (i.e. atom-typing rules) [Citation31,Citation64] along with the associated force field parameters. While there are freely available tools to aid in atom-typing [Citation65–69], these are typically specific to a particular force field or simulator, and/or capture the atom-typing and parametrization in a hierarchy (either through specific placement in a parameter file read by the code or as nested if/else statements within the source code). Foyer does not encode usage rules into the source code, instead defining usage rules and parameters in an XML file that is an extension of the OpenMM [Citation70] force field file format. The Foyer software itself is used to interpret and apply the rules and thus the software is not limited to use with only a single force field type. By separating the usage rules from the source code, changes or extensions to force field parameters/rules does not require changes to the code itself. Force field usage rules are encoded using a combination of a SMARTS-based [Citation71] annotation scheme, which defines the molecular environment (i.e. chemical context) associated with a given parameter, and overrides that define rule precedence (i.e. which atom type to choose when multiple rules can apply to an interaction site). The use of overrides avoids the need to define rule precedence via the order of the rules within a file (See [Citation64] for more details). As an example, Listing 1 shows the contents of an XML file that contains parameters and usage rules from the OPLS force field for linear alkanes. We note that Foyer allows user-defined input (by pre-pending with an underscore), allowing SMARTS to be used for non-elemental interaction sites (e.g. an interaction site that represents a coarse-grained bead or an united atom interaction site). As such, the exact parameters and their usage can be readily captured and disseminated along with a simulation and/or publication. This provides an improved way to disseminate custom force field parameter sets and/or novel force field parameters (e.g. see Ref. [Citation32]) that reduces ambiguity, as the format used by Foyer to encode the usage rules and parameters is both human and machine readable; thus parameterisation rules provided in a publication can be automatically tested for accuracy and completeness. To further enhance reproducibility, the XML force field files additionally include a doi tag for the source of the parameters (see Listing 1); upon successful atom-typing, Foyer outputs a BibTeX file of references with the relevant DOIs, significantly improving the transparency as to the origin of parameters used in a simulation and therefore reproducibility.
2.2. Other community tools
Here we briefly highlight other simulation tools and efforts with a considerable focus on reproducibility and transparency, several with similar and/or complementary functionality to MoSDeF. We do not include discussion of commercial tools, as the need to purchase software places a fundamental roadblock in terms of reproducibility.
The Atomic Simulation Environment (ASE) [Citation72] is a Python toolkit that provides wrappers to various programmes/libraries allowing atomistic simulations to be setup, run and analysed within a single script. Support is provided for numerous electronic structure codes and several MD simulation engines; however, as ASE is primarily focussed on hard matter systems it does not currently support robust tools for initialisation of complex soft matter systems or atom-typing.
Pysimm [Citation73,Citation74], is an open-source Python toolkit for soft matter systems providing routines for system setup and wrappers that support LAMMPS MD [Citation61] and Cassandra MC [Citation44] engines, allowing a simulation workflow to be encoded in a Python script. Of particular note, pysimm includes routines that simplify the process for performing complex workflows such as simulated growth/crosslinking of polymers [Citation75]. We note that since both ASE and pysimm are also developed as Python libraries, there is a natural level of interoperability between these tools and MoSDeF. Hoobas is another open-source molecular building package that facilitates the construction of polymers for molecular dynamics simulation [Citation76,Citation77]. indigox is an open-source package that utilises the CherryPicker algorithm to help parametrise molecules based on fragments of previously-parametrised molecules [Citation78]. Open Babel is a library of cheminformatics functions that support constructing molecular models, SMARTS-matching, and basic molecular dynamics functions with basic molecular mechanics force fields [Citation79,Citation80]. OpenKIM is a multifaceted toolkit providing a portal for storage of interatomic models and their associated data, and an application programming interface (API) created such that models can work seamlessly (and correctly) between different simulation engines; we note this API is designed to ensure parameters are defined correctly, not to perform atom-typing or to encode usage rules and does not provide tools for system initialisation or workflow management. To date, OpenKIM has largely focussed on atomic systems (i.e. a system is defined solely by its atoms, and ‘bonds’ are an outcome of atomic positions), whereas most soft-matter force fields include both non-bonded and bonded parameters and assign different parameters to atoms based on the bonds. The Open Force Field consortium [Citation81–83] has developed a variety of open-source tools that utilise chemical perception via SMIRKS (<https://www.daylight.com/dayhtml/doc/theory/theory.smirks.html>) patterns to identify atom types and other force field parameters pertinent to each atom in a chemical system, similar to Foyer's underlying methodology. WebFF is an ongoing NIST-project aimed at developing an infrastructure for modelling soft materials and curating force field data for traceable data provenance [Citation84]. BioSimSpace provides an API that allows users to mix-and-match various molecular modelling tools, facilitating the use of complex workflows involving molecular dynamics, metadynamics, various water models, various force fields, free energy methods, and various simulation engines [Citation85]. signac is a Python library that provides basic components required to create a well-defined and collectively accessible data space and enables data access and modification through a homogeneous data interface that is agnostic to the data source. signac-flow is an extension of the signac framework [Citation34], which aids in the management of highly complex data spaces. signac-flow allows submission to high performance computing (HPC) scheduling systems, including both PBS and SLURM. Since signac-flow captures the entire workflow definition and execution, it can be used to facilitate reproducible workflows. mBuild and Foyer have been used in combination with signac-flow in several past and on-going research projects by the authors [Citation7,Citation8]. FireWorks is another workflow manager that supports dynamic workflows using MongoDB [Citation86,Citation87].
3. TRUE molecular simulations
We have defined TRUE molecular simulations as ones that are transparent, reproducible, usable by others and extensible. In this section, we provide some examples of TRUE simulations utilising the capabilities of MoSDeF. Fully detailed instructions for installing MoSDeF and the related tools needed to run each example are contained within the Supporting Information. But first we define what we mean by these terms in the context of molecular simulation.
A simulation is transparent when all the information needed to exactly follow the steps undertaken by the original author(s) (such as all scripts used to set up the system, details of force field implementation, all input files to the simulation engines, any other needed input files) are visible to anyone in the community. This requires the sharing of this information in a version-controlled persistent open-source repository, such as GitHub. This information, in and of itself, may only be useful to a true expert; however, few simulations published today meet even this standard. A transparent simulation is reproducible when sufficient information (in supplementary information and/or documentation) is provided so that future researchers interested in duplicating the work can construct and run the reported simulation. From this point of view, a self-contained workflow - such as a Jupyter notebook, or a virtual machine - is highly desirable. As defined here, reproducibility does not require a high level of expertise - for example, the calculation could be reproduced by a student in a class, a newcomer to simulation, etc. In particular, Jupyter notebooks provide a convenient, high-level representation of a script that integrates with other common Python tools and can be converted directly into a Python script using nbconvert. Two caveats about reproducibility must be borne in mind. First, we note that in molecular simulation, reproducibility is unlikely to be exact, in the following sense: Two MD simulations, when run on different architecture machines, will not generate the same trajectory due to differences in the handling of floating point operations. As in any nonlinear dynamical system, small differences between trajectories (due to different rounding errors) grow exponentially large over time. Even when run on the same computer, two simulations may not give the exact same trajectory. This is because of parallelised computing, in which parts of the calculation are done by separate processors and then gathered (added together) in an order that is not predictable due to fluctuations in message passing times. The problem is exacerbated even more in MC simulations, where a difference in random number seed will generate a different sequence of random numbers on the same machine with the same random number generator. On different machines, trying to achieve reproducibility in MC simulations at the level of configurations on different machines requires using the same random number generator with reproducible arithmetic (IEEE standard-compliant) with the same seed; additionally, the same issue with parallelisation noted for MD simulations also applies [Citation88]. However, we do not expect reproducibility at the level of individual simulation trajectories; what we expect is statistical reproducibility in the averages of the properties calculated over the course of the simulation. Second, many simulations that are reported in the literature require prodigious amounts of computational resources, such as millions of hours on a leadership-class supercomputer. In this case, having available all of the codes means that reproducibility is limited to those having available to them similar levels of computing resources. In this case, we propose that researchers may also elect to make available a simplified version of the reported calculation accessible to those that have limited computational resources (for example, using a much smaller system size and shorter simulation times or a single physiochemical statepoint instead of many). Such scaled-down versions could also have considerable pedagogical value.
We define a transparent, reproducible simulation to be usable by others when a future researcher can utilise the available files and documentation to reproduce the calculation and make use of the data generated - for example, to analyse the trajectory/trajectories for different properties. This requires a level of documentation that includes information about where output files are located in the data space created by reproducing the simulation, and how these files might be analysed in different ways. Finally, a transparent, reproducible, usable-by-others simulation is extensible if the documentation is sufficiently detailed that a future researcher could change characteristics of the simulation - such as changing molecular species, state conditions, simulation engine, properties calculated, etc.
By adhering to the principles of TRUE simulations, researchers will enable their work to be utilised in ways that have not been hitherto possible. In particular, it will create resources that lower the barrier to entry into the field of molecular simulation, as well as allow researchers to distribute their research in a more useful fashion. Using MoSDeF is not necessary to create TRUE simulations, but as the examples below illustrate, MoSDeF makes it considerably easier to distribute TRUE simulations by automating and standardising many of the steps, thus minimising the documentation needed to create a TRUE simulation. Also, the open-source nature of MoSDeF offers the ability for researchers to make contributions to the code base in the form of methods, recipes, force fields, etc.
3.1. Ethane VLE using TraPPE
One popular application of molecular simulation involves the use of Monte Carlo (MC) methods, often employing extended ensembles in techniques such as Gibbs Ensemble Monte Carlo (GEMC) or grand canonical Monte Carlo (GCMC), to simulate vapour-liquid equilibria (VLE) properties. Briefly, GEMC involves simulating two distinct simulation boxes (which generally have different densities and compositions) and performing MC moves to perturb both systems to balance the chemical potentials and pressures between the two simulation boxes [Citation89,Citation90], thus reaching phase equilibrium. This involves particle displacements within boxes, particle exchanges across boxes, and box volume changes [Citation89,Citation90]. GCMC methods, on the other hand, involve simulating a single simulation box, but performing MC moves to insert or delete particles from a reservoir [Citation91]. Additionally, more complex MC moves have been proposed to accelerate equilibration for systems containing complex molecules, including configurational bias Monte Carlo (CBMC) methods [Citation92]. The transferable potentials for phase equilibria (TraPPE) force field has been designed for conducting simulations for phase equilibria [Citation93,Citation94]. Here, we present a TRUE workflow that examines ethane vapour-liquid coexistence at a single thermodynamic statepoint. This workflow utilises mBuild [Citation28,Citation29] to initialise two simulation boxes of ethane (vapor and liquid phases), Foyer [Citation30,Citation64] to apply the TraPPE-United Atom (TraPPE-UA) force field [Citation93], and GOMC [Citation56,Citation57,Citation95] to perform a GEMC simulation. mBuild is used to pack ethane into two different simulation boxes according to the vapour and liquid densities at 236 K (Figure ).
Figure 3. Two boxes of ethane constructed in mBuild. Liquid phase (left) and vapour phase (right) are simulated simultaneously in GEMC.

Foyer is used to systematically apply TraPPE force field parameters. GOMC (version 2.40) is used to perform the GEMC simulation at 236 K, with simulation parameters consistent with the TraPPE implementation [Citation93,Citation94]: Lorentz-Berthelot combining rules, fixed bonds, 1.4 nm Lennard-Jones cutoffs, analytical tail corrections, and Ewald summations for electrostatic interactions. The resultant analysis validates the vapour pressure, vapour density, and liquid density at 236 K against published reference data (Figure ) [Citation93,Citation94]. A link to this GitHub repository can be found in the Supporting Information. All Python dependencies related to building, simulating, and analysing are openly available and well-documented, and routines are built on top of these dependencies that expose chemistry and statepoint variables.
Figure 4. Vapor pressure (left), vapour density (middle), and liquid density (right) plots for ethane at 236 K, using GEMC in GOMC with the TraPPE force field.
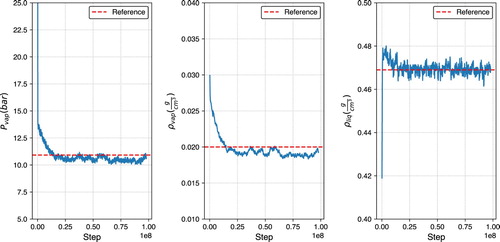
This workflow is transparent and reproducible, as this workflow and relevant software packages are open-source and available on GitHub [Citation29,Citation30,Citation57,Citation96]. Furthermore, the workflow is usable by others, as the logged quantities can be analysed for other properties beyond vapour pressure and densities. Lastly, this workflow is extensible, as there is a pattern and clear room to implement other state points, molecules, force fields, or simulation engines in addition to implementing workflow managers to facilitate large-scale screening studies.
3.2. Graphene slit pore
Graphene has been extensively researched as an electrode material for energy storage applications [Citation97–99] in recent years mainly due to its high surface area [Citation97,Citation98,Citation100]. Furthermore, the interactions between graphene pores and fluid molecules were studied with MD simulations through the use of slit pore models [Citation101,Citation102]. Here we demonstrate a TRUE simulation workflow for a graphene slit pore solvated with aqueous NaCl. This TRUE graphene simulation was performed with the use of mBuild [Citation28,Citation29], Foyer [Citation30,Citation64], GROMACS [Citation38–40,Citation103–105], and MDTraj [Citation106]. Pore-Builder [Citation107], an mBuild ‘recipe’, was also used to initialise the graphene sheets contained in the system.
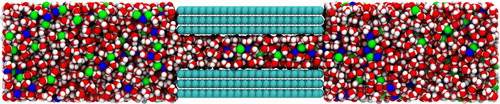
In this specific system, a graphene slit pore filled with aqueous NaCl, was initialised with mBuild. mBuild Compounds of the specific molecules were initialised with the mbuild.load() function using MOL2 files. Once the molecule Compounds were initialised, the GraphenePoreSolvent class within Pore-Builder was utilised. This specific class makes use of the mbuild.Lattice class and the mbuild.solvate function to build a graphene slit pore system solvated with fluid specified by the user. In this system, the graphene slit pore was built with three sheets on each side, and a pore width of 1.5 nm. Additionally, the length of the graphene sheet in the x direction was set to 5 nm and the length of the graphene sheet in the z direction was set to 4 nm. The bulk region of fluid was set to 6 nm on each side of the slit pore. 5200 waters and 400 Na and Cl ions were solvated into the system. A snapshot of the system is shown in Figure .
Figure 5. A snapshot of the graphene slit pore system containing graphene carbon (cyan), water (red for oxygen and white for hydrogen), sodium ions (blue) and chlorine ions (green).
Once the graphene slit pore system was initialised as an mBuild compound, it was atom-typed and parametrised with Foyer. Three force fields were used in this system, with their information stored in three separate XML files: GAFF [Citation11], SPC/E [Citation108], and the force field of Joung and Cheatham (JC) [Citation109]. GAFF describes the interactions between the graphene atoms, SPC/E describes the water interactions, and JC describes the Na and Cl interactions. Each force field uses 12-6 Lennard-Jones interactions, point charges, and harmonic bonds and angles.
The simulation was run with GROMACS 2018.5. Steepest descent energy minimisation was first performed for 1000 steps to remove any energetic clashes from the initial configuration. Afterwards, a series of two MD simulations were performed with the following parameters: cutoffs of 1.4 nm for Coulombic and van der Waals interactions, a temperature of 300 K controlled with the v-rescale thermostat with a time constant of 0.1 ps, particle mesh Ewald to handle long-range electrostatics, and a timestep of 1 fs. Additionally, the graphene atoms were frozen in place. A GROMACS NDX file was created with a Water_and_ions group so that the thermostat could be applied to the fluids; no thermostat is applied to the graphene, as the graphene is kept rigid. First NVT equilibration was performed to further relax the system of any unfavourable configurations for 100,000 steps. Afterwards, NVT sampling was performed for 2,500,000 steps. In the sampling run, all bonds were constrained using the LINCS [Citation110] algorithm.
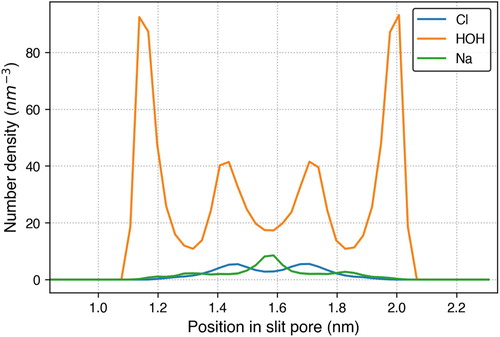
Once the sampling simulation was performed, the number density profile of each fluid type is calculated with the use of MDTraj and plotted with Matplotlib. The results are shown in Figure . From these results, we observe that the water molecules are mainly structured near the pore walls at 1.2 and 2.0 nm. Additionally, there are two smaller peaks around 1.4 and 1.8 nm indicating structuring of water in the middle of the pore. The Na ions are structured in the middle of the pore around 1.6 nm and the the Cl ions are structured to each side of the Na ions, at around 1.5 and 1.7 nm. If the graphene was positively or negatively charged, we would expect different structure behaviour of the ions. This simulation can be extended to further understand the effect of various parameters on the fluid structure within the pore. For example, the user can easily specify a different pore width to study how this impacts the structure of water and ions. This workflow is encapsulated in a Jupyter notebook, providing the user access to modify any of these high-level parameters.
Figure 6. Number density profiles across the width of the pore for water, Na, and Cl.
The workflow for simulating a graphene slit pore satisfies the conditions to be a TRUE simulation. First, this workflow is transparent as all scripts, input files, and force field information are available for anyone to view [Citation111]. Next, this workflow is reproducible as the exact steps to set up and run the simulation are contained within a Jupyter notebook. Barring differences in computer architectures and parallelisation schemes, a user running this Jupyter notebook should be able to reproduce the number density profiles from the reference simulation. Additionally, the trajectories are kept within the workflow directory, allowing users to analyse properties other than number density. Finally, the functions and classes used to initialise the graphene slit pore system are sufficiently documented so that a user may change characteristics of the simulation if they wish. For example, a user can extend this workflow to study additional aqueous solutions.
3.3. Lipid bilayers
MD is a common technique used to perform simulation of biological systems. An example TRUE biological simulation workflow is demonstrated in the true_lipids repository on GitHub [Citation112]. This workflow focuses on simulating lipids found in the outermost layer of the skin, the stratum corneum (SC). The SC, which is primarily composed of ceramides (CER), cholesterol (CHOL), and free fatty acids (FFA) [Citation113], essentially controls the barrier function of the skin [Citation114]. In this workflow a hydrated pre-assembled bilayer configuration of skin lipids was initialised, simulated, and analysed in a well-documented and reproducible fashion.
mBuild was used to initialise the system configuration, specifically utilising the Bilayer [Citation115] recipe. A simplified model system containing only CER N-hydroxysphingosine (NS) C24:0, CHOL, and FFA C24:0 was chosen for this example; however a more complex mixture could be easily built by the Bilayer recipe. For each leaflet of the bilayer, 36 lipids were randomly placed on a lattice and rotated about the bilayer normal axis. The lattice was set up and spaced such that the lateral area occupied by each lipid was equal to the target and as designated by the area_per_lipid parameter. In addition, the lipids were rotated about a randomly chosen axis parallel to the bilayer by the tunable tilt_angle parameter. Finally, 20 waters per lipid were added to each of the two ends of the simulation box at a density of
. The full system contains 72 lipids and 2880 water molecules. While many of the steps involved in setting up the initial configuration involve random number generators, exact reproducibility on the same machine was enforced by the initialisation of a random seed.
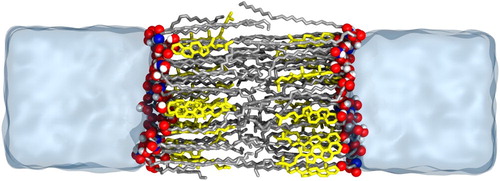
Simulations were conducted using the GROMACS 2018.5 [Citation38–40,Citation103–105] MD engine, using a modified CHARMM36 force field [Citation116,Citation117] with a 1 fs timestep. The system was first energy-minimised using the steepest descent algorithm for 20,000 steps in order to remove high energy atomic contacts. Temperature fluctuations were stabilised by running a 500 ps NVT simulation using the Berendsen thermostat [Citation118] at 305 K with a time constant of 1 ps. Next, the volume fluctuations were stabilised with a 10 ns NPT simulation at 305 K and 1 atm. This step and all others hereinafter in this section were in the NPT ensemble and use the Nosé-Hoover thermostat [Citation119] with a time constant of 1 ps and the Parinello-Rahman barostat [Citation120] with a time constant of 10 ps and a compressibility of . Still at 1 atm, the system was linearly annealed to 340 K over 5 ns, held at 340 K for 15 ns, linearly cooled to 305 K over 5 ns, and held at 305 K for 25 ns in order to accelerate equilibration of the rotational orientation of the lipids. Finally, the system was run for 20 ns at 305 K and 1 atm, saving coordinates to a trajectory file every 10 ps. The final snapshot of the system is shown in Figure . More details on the simulation parameters can be found in the Supporting Information.
Figure 7. Simulation snapshot of lipid bilayer containing CER N-hydroxysphingosine C24:0 (CER NS), cholesterol and lignoceric acid. The CER NS and FFA tails are shown in silver, cholesterol in yellow, and the headgroup oxygen, nitrogen and hydrogen atoms in red, blue and white respectively.
The trajectory from the final step was analysed using MDTraj [Citation106]. Neutron scattering is a popular tool to experimentally obtain structural information of lipid lamella. A neutron scattering length density (NSLD) profile was calculated for this simulated system along the bilayer normal axis in Figure .
Figure 8. Simulated NSLD profiles for specifically deuterated lipid tails.
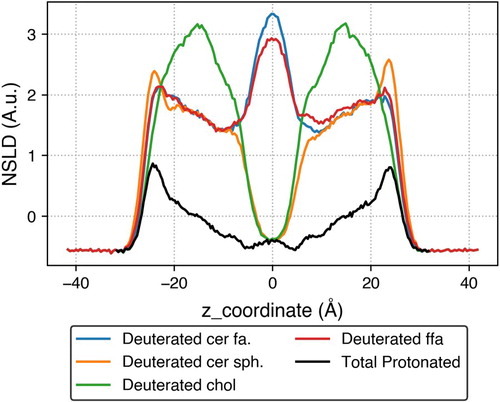
It is apparent from these profiles that the 24-carbon fatty acid tail of the CER and the 24-carbon FFA tail interdigitate, as indicated by the high density peak in the centre of the profile. One can also observe that the 16-carbon sphingosine tail of the CER and CHOL do not interdigitate, and are not present in the middle of the bilayer as there is a low-density trough in their deuteration profiles. The scattering length densities at the outer edges of the bilayer suggest that the CHOL headgroup is located closer to the centre of the bilayer compared to that of other lipids. In addition to the NSLD profiles, an area per lipid of , a tail tilt angle
, a nematic order parameter of 0.9414 and a bilayer height of
were calculated in the workflow.
All of these values and plots can be reproduced by executing the workflow. Furthermore, by extending the workflow to screen over the parameter space, one could identify trends in the calculated values. The Bilayer recipe is highly modular allowing the user to easily create reproducible bilayer structures containing different lipid types, system sizes, compositions, or water content using an intuitive Python script.
3.4. Friction reduction via thin film coatings
Thin film coatings can be used to modify the surface properties of different systems [Citation121]. One potential application of these coatings is to improve tribological properties of surfaces at the micro and nanoscale [Citation36,Citation121,Citation122]. In this example, we present a TRUE simulation of a thin film coated system, which is based on an in-depth study by Summers et al. [Citation36]. Specifically, we built a system of two rectangular silica surfaces, parallel to one another. Each surface was coated with 100, 17-carbon long, alkylsilane chains, all of which were terminated with a methyl group. Surface oxygens not functionalised with chains were backfilled with hydrogen caps to emulate protonated hydrolysis. These systems were created with mBuild [Citation28,Citation29], and atom-typed, parametrised with Foyer [Citation30,Citation64] using OPLS-aa [Citation10] force field parameters. A visualisation of the described system is presented in Figure .
Figure 9. Thin film coated surfaces model.

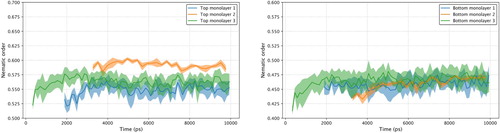
The system was simulated with LAMMPS [Citation123] and GROMACS [Citation38–40,Citation103–105]. MD simulations were run under the canonical ensemble (NVT) and a Nosé-Hoover thermostat maintaining the temperature at 298 K [Citation119]. Long-range electrostatics were calculated using the particle-particle particle-mesh (PPPM) algorithm [Citation124]. The rRESPA time step algorithm was utilised with 0.25 fs, 0.5 fs, 0.5 fs, and 1.0 fs timesteps for bonds, angles, dihedrals, and non-bonded interactions, respectively [Citation125]. The simulation started with energy minimisation with LAMMPS for 10,000 steps, followed by another 50,000 steps with GROMACS to bring the monolayers to a more relaxed state. This process continued with NVT equilibration by GROMACS to bring the system to a desired stable state for 1,000,000 steps. The workflow proceeded to use GROMACS to compress the system by pulling the top surfaces along the z axis to come into contact with the bottom surface. In the next step, the top surface was sheared against the bottom surface by imposing a force to pull it along the z axis at the rate of 0.01 nm/ps. The production run was designed to simulate for 5,000,000 steps, which would be equivalent to 10 ns of shearing. From the GROMACS output file, the properties of the system can be calculated. The steady-state production period used for final data analysis was determined using the automatic equilibration detection method provided by pymbar [Citation126,Citation127]. By using a defined method to determine the steady-state cutoff, the consistency of the data analysis process can be ensured, holding to the first two criteria of TRUE, transparent and reproducible. The calculated nematic order of three example runs were determined and are presented in Figure .
Figure 10. Steady state nematic order of the thin film coated on top and bottom surfaces.
This example focuses on showcasing the extensibility of TRUE, emphasising the ability to modify and expand the project beyond the original study and parameters of interest. This goal can be achieved by applying Object-Oriented Programming (OOP) design principles, in combination with open-source libraries. Encapsulating frequently used code into classes and functions helps improve the reusability of codes and make it easier to create novel systems, just by changing and adding new variables. By pairing MoSDeF suite libraries, mBuild [Citation28,Citation29] and Foyer [Citation30,Citation64], with other open-source libraries, such as signac and signac-flow, part of the signac framework [Citation33,Citation128], the extensibility could be made even more manageable and achievable. Most importantly, all building blocks of the project have to be properly documented, either as comments in the code or in a separate manual. These practices can help projects expand effectively. For instance, in this example, although the arguments and variables were defined such that the surfaces were filled with 100, 17-carbon long, alkylsilane chains, each then capped with a methyl group, many unique systems can be created by altering the chain density, backbone chain length, backbone chemistries, terminal groups, and others as need arises. The latter part of the example shows how we can expand the project from the original system by varying backbone chain lengths. For the sake of demonstration, we only show the first few steps of the workflow, starting with setting up the workspace using signac [Citation33,Citation128], building corresponding systems with mBuild [Citation28,Citation29], and atom-typing, parametrising with Foyer [Citation30,Citation64]. Other steps of the simulation can be added analogously. We implement signac-flow [Citation33,Citation128] as the workflow manager. These tools will become vital when needing to run a complete workflow and managing thousands of systems. All scripts and files needed to run the above example are located in a GitHub repository [Citation129]. Users can interface with this example through the Jupyter notebook located within the repository. By providing properly documented codes and scripts used to set up the system, using open-source libraries to perform simulation and data analysis, the first three criteria of TRUE are also satisfied. This example workflow is Transparent, Reproducible, Usable by others, and Extensible, or concisely, a TRUE simulation.
4. Conclusions
In this paper, we have outlined some of the key issues related to reproducibility in molecular simulations of soft matter. We have also discussed many practices that computational scientists could implement in efforts to result in more reproducible science, such as using scripts instead of manual input, using open-source software tools, and using version control and modern software development practices when developing software. In this paper, we assert three central claims:
The goal in computational molecular science should be simulations that are TRUE: Transparent, Reproducible, Usable by others, and Extensible.
Scientific results reported in the literature that depend on molecular simulations should adhere to the above characteristics.
Use of the Molecular Simulation and Design Framework (MoSDeF) is one way to enable TRUE simulations.
To demonstrate the third claim, we revisit some ‘ten rules’ papers [Citation20–22] that provide succinct instructions for practicing reproducible science and demonstrate how the above example workflows utilise MoSDeF to this end. A common recommendation in these discussions is that every step in a workflow should be automated and free of manual input, i.e. scriptable. MoSDeF, in its current state, is a set of Python libraries designed specifically to address this. In a single Python script (or Jupyter notebook), each step of a molecular simulation workflow (generation of particle coordinates, application of a force field, running of a molecular simulation, and analysis of the results) can be specified and run. The objective of measuring physical properties from some chemical input can be achieved with one call to an executable (although the simulation may take some hours or days to run). In order for these scripts, which include many imports to other Python libraries, to produce identical (or sufficiently identical) results some years in the future, the underlying libraries must be version-controlled. The core MoSDeF packages (mBuild and Foyer) undergo regular releases, tagged with semantic version numbers, every few weeks or months as they are developed. Other packages, such as simulation engines, the packages in the signac framework, and underlying scientific Python packages, are also version-controlled and undergo regular releases. Specifying the version of each software package used in a simulation workflow is not necessarily sufficient to ensure reproducible science, but it is a significant improvement over the use of ad hoc or in-house scripts that often lack version control, proper testing, or releases. Similarly, it has been argued that the use of community-developed software libraries, and the extension of such libraries, further promotes reproducibility as compared to closed-source, in-house development [Citation19]. MoSDeF is a set of open-source that interface with other open-source, community-developed libraries and software tools.
Additionally, MoSDeF makes use of virtually no GUIs - or, more specifically, no GUIs that hide the details of a simulations protocol from the user. Some molecular visualisation tools (NGLview, py3DMol, VMD, ovito, fresnel) can be used in conjunction with MoSDeF, but these are only tools to visualise systems and do not hide workflow details or replace steps in a workflow.
Finally, we would like to discuss an additional benefit of shifting toward more reproducible computational studies: the facilitation of large-scale screening of physiochemical space. Continuous improvements in computer hardware and recent advancements in machine learning methodologies have driven interest in studying large data sets, typically many orders of magnitude larger than typically seen in the literature. Provided that each step in a workflow can be automated - in other words, scriptable with no manual input - a single simulation can be repeated with different physical inputs (e.g. at different thermodynamic statepoints or with different chemistries) by only modifying the input parameters. For example, consider some system at temperature and pressure for which we care about some physical property A. One can run a simulation at
and get property
but later decide we want to look at some other temperature and/or pressure. One could manually move some files around and get property
from statepoint
without prohibitive trouble, but doing this once is a plausible source of human error and repeating this process many times is not feasible. Screening over N statepoints in a reproducible manner necessitates that running a workflow at a single statepoint is reproducible. We hope the practices outlined in this paper and the use of MoSDeF can enable reproducible computational science at each scale.
Supplemental Material
Download PDF (154.4 KB)Acknowledgements
We acknowledge key contributions to MoSDeF by former group members Christoph Klein (myriad.com) and Andrew Z. Summers (enthought.com) and by ISIS staff Peter Volgesi, Umesh Timalsina, and Janos Sallai.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Related Research Data
References
- M. Baker, Nature 533 (7604), 452–454 (2016).
- E.B. Tadmor, R.S. Elliott, S.R. Phillpot and S.B. Sinnott, Curr. Opin. Solid State Mater. Sci. 17 (6), 298–304 (2013).
- A. Jain, S.P. Ong, G. Hautier, W. Chen, W.D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K.A. Persson, APL Mater. 1 (1), 11002 (2013).
- C.E. Wilmer, M. Leaf, C.Y. Lee, O.K. Farha, B.G. Hauser, J.T. Hupp and R.Q. Snurr, Nat. Chem. 4 (2), 83–89 (2012).
- J. Hachmann, R. Olivares-Amaya, S. Atahan-Evrenk, C. Amador-Bedolla, R.S. Sanchez-Carrera, A. Gold-Parker, L. Vogt, A.M. Brockway and A. Aspuru-Guzik, J. Phys. Chem. Lett. 2 (17), 2241–2251 (2011).
- M.A.F. Afzal, M. Haghighatlari, S.P. Ganesh, C. Cheng and J. Hachmann, J. Phys. Chem. C 123 (23), 14610–14618 (2019).
- M.W. Thompson, R. Matsumoto, R.L. Sacci, N.C. Sanders and P.T. Cummings, J. Phys. Chem. B123 (6), 1340–1347 (2019).
- R.A. Matsumoto, M.W. Thompson and P.T. Cummings, J. Phys. Chem. B 123, 9944 (2019). <https://doi.org/10.1021/acs.jpcb.9b08509>.
- OSTP, Materials Genome Initiative for Global Competitiveness, 2011.
- W.L. Jorgensen, D.S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc. 118 (45), 11225–11236 (1996).
- J. Wang, R.M. Wolf, J.W. Caldwell, P.A. Kollman and D.A. Case, J. Comput. Chem. 25 (9), 1157–1174 (2004).
- S. Jo, T. Kim and W. Im, PLoS ONE 2 (9), e880 (2007).
- M. Schappals, A. Mecklenfeld, L. Kröger, V. Botan, A. Köster, S. Stephan, E.J. García, G. Rutkai, G. Raabe, P. Klein, K. Leonhard, C.W. Glass, J. Lenhard, J. Vrabec and H. Hasse, J. Chem. Theory Comput. 13 (9), 4270–4280 (2017).
- B. Chen, J.I. Siepmann, S. Karaborni and M.L. Klein, J. Phys. Chem. B 107 (44), 12320–12323 (2003).
- F.M.S. Silva Fernandes, F.F.M. Freitas and R.P.S. Fartaria, J. Phys. Chem. B 108 (26), 9251–9255 (2004).
- J.C. Palmer, A. Haji-Akbari, R.S. Singh, F. Martelli, R. Car, A.Z. Panagiotopoulos and P.G. Debenedetti, J. Chem. Phys. 148 (13), 137101 (2018).
- A.G. Smart, The War Over Supercooled Water American Institute of Physics, August 2018. <https://physicstoday.scitation.org/do/10.1063/PT.6.1.20180822a/full>.
- S. Jo, T. Kim, V.G. Iyer and W. Im, J. Comput. Chem. 29 (11), 1859–1865 (2008).
- D.L. Donoho, A. Maleki, I.U. Rahman, M. Shahram and V. Stodden, Comput. Sci. Eng. 11 (1), 8–18 (2009).
- G.K. Sandve, A. Nekrutenko, J. Taylor and E. Hovig, PLoS Comput. Biol. 9 (10), e1003285 (2013).
- A. Elofsson, B. Hess, E. Lindahl, A. Onufriev, D. van der Spoel and A. Wallqvist, PLoS Comput. Biol. 15 (1), 2–5 (2019).
- L.A. Barba, Science 354 (6308), 142–142 (2016).
- L.A. Barba, Lorena A. Barba Blog. <http://lorenabarba.com/category/blog/>.
- C.M. Reinhart and K.S. Rogoff, Am. Econ. Rev. 100 (2), 573–578 (2010).
- T. Herndon, M. Ash and R. Pollin, Cambridge J. Econ. 38 (2), 257–279 (2014).
- MoSDeF Web Site. <http://www.mosdef.org>.
- Institute for Software Integrated Systems. <https://www.isis.vanderbilt.edu/>.
- C. Klein, J. Sallai, T.J. Jones, C.R. Iacovella, C. McCabe and P.T. Cummings, in Foundations of Molecular Modeling and Simulation. Molecular Modeling and Simulation (Applications and Perspectives), edited by Randall Q. Snurr, Claire S. Adjiman and David A. Kofke (Springer, Singapore, 2016), pp. 79–92.
- mBuild Github Repository. <https://github.com/mosdef-hub/mbuild>.
- Foyer Github Repository. <https://github.com/mosdef-hub/foyer>.
- C.R. Iacovella, J. Sallai, C. Klein and T. Ma, Idea Paper: Development of a Software Framework for Formalizing, 2016.
- J.E. Black, G.M.C. Silva, C. Klein, C.R. Iacovella, P. Morgado, L.F.G. Martins, E.J.M. Filipe and C. McCabe, J. Phys. Chem. B 121 (27), 6588–6600 (2017).
- C.S. Adorf, P.M. Dodd, V. Ramasubramani and S.C. Glotzer, Comput. Mater. Sci. 146, 220–229 (2018).
- Signac Documentation. <https://signac.readthedocs.io/en/latest/>.
- A.Z. Summers, C.R. Iacovella, P.T. Cummings and C. McCabe, Langmuir 33 (42), 11270–11280 (2017).
- A.Z. Summers, J.B. Gilmer, C.R. Iacovella, P.T. Cummings, and C. McCabe, J. Chem. Theory Comput. 16, 1779–1793 (2020. PMID: 32004433. DOI:10.1021/acs.jctc.9b01183.
- MoSDeF-Anaconda Cloud. <https://anaconda.org/mosdef/>.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput. 4 (3), 435–447 (2008).
- H.J.C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun. 91 (1), 43–56 (1995).
- M.J. Abraham, T. Murtola, R. Schulz, S. Páall, J.C. Smith, B. Hess and E. Lindah, SoftwareX1-2, 19–25 (2015).
- S. Plimpton, J. Comput. Phys. 117 (1), 1–19 (1995).
- J.A. Anderson and A. Travesset, Comput. Sci. Eng. 227 (10), 5342–5349 (2008).
- HOOMD-blue (2017). <http://glotzerlab.engin.umich.edu/hoomd-blue>.
- J.K. Shah, E. Marin-Rimoldi, R.G. Mullen, B.P. Keene, S. Khan, A.S. Paluch, N. Rai, L.L. Romanielo, T.W. Rosch, B. Yoo and E.J. Maginn, J. Comput. Chem. 38 (19), 1727–1739 (2017).
- ParmEd – ParmEd Documentation (February 2018). <http://parmed.github.io/ParmEd/html/index.html>.
- J. Sztipanovits and G. Karsai, Computer 30 (4), 110–111 (1997).
- C.R. Iacovella, G. Varga, J. Sallai, S. Mukherjee, A. Ledeczi and P.T. Cummings, Theor. Chem. Acc. 132 (1), 1315 (2013).
- Travis CI: A Hosted Continuous Integration Service. <http://travis-ci.org>.
- Azure Pipelines Landing Page. <https://azure.microsoft.com/en-us/services/devops/pipelines/>.
- CodeCov Landing Page. <https://codecov.io/>.
- Coveralls Landing Page. <https://coveralls.io>.
- Slack Virtual Shared Workplace Tool. <https://slack.com>.
- M. Davenport, Chem. Eng. News 94 (29), 23–24 (2016).
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph. 14 (1), 33–38 (1996).
- Collaborative Research: NSCI Framework: Software for Building a Community-Based Molecular Modeling Capability Around the Molecular Simulation Design Framework (MoSDeF). <https://www.nsf.gov/awardsearch/showAward?AWD_ID=1835874>.
- GPU-Optimized Monte Carlo (GOMC) Home Page (April 2018). <http://gomc.eng.wayne.edu/>.
- GOMC, GPU-Optimized Monte Carlo (GOMC) Github Repository (April 2018). <https://github.com/GOMC-WSU/GOMC>.
- CP2K Open Source Molecular Dynamics (April 2018). <https://www.cp2k.org/>.
- Signac-flow Webpage. <http://signac-flow.readthedocs.io>.
- R. Salomon-Ferrer, D.A. Case and R.C. Walker, Wiley Interdiscip. Rev. Comput. Mol. Sci. 3 (2), 198–210 (2013).
- S. Plimpton, M. Jones and P. Crozier, Pizza.py Toolkit Sandia National Laboratories (February 2006). <http://pizza.sandia.gov/>.
- A.I. Jewett, Z. Zhuang and J.E. Shea, Biophys. J. 104 (2), 169a (2013).
- L. Martínez, R. Andrade, E.G. Birgin and J.M. Martínez, J. Comput. Chem. 30 (13), 2157–2164 (2009).
- C. Klein, A.Z. Summers, M.W. Thompson, J.B. Gilmer, C. McCabe, P.T. Cummings, J. Sallai and C.R. Iacovella, Comput. Mater. Sci. 167, 215–227 (2019).
- J. Wang, W. Wang, P.A. Kollman and D.A. Case, J. Mol. Graph. Model. 25 (2), 247–260 (2006).
- K. Vanommeslaeghe and A.D. MacKerell, Jr., J. Chem. Inf. Model. 52 (12), 3144–3154 (2012).
- A.K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P.C. Nair, C. Oostenbrink and A.E. Mark, J. Chem. Theory Comput. 7 (12), 4026–4037 (2011).
- A.A.S.T. Ribeiro, B.A.C. Horta and R.B. de Alencastro, J. Braz. Chem. Soc. 19 (7), 1433–1435 (2008).
- B.L. Eggimann, A.J. Sunnarborg, H.D. Stern, A.P. Bliss and J.I. Siepmann, Mol. Simul. 40 (1–3), 101–105 (2014).
- SimTK: OpenMM: Project Home (February 2018). <https://simtk.org/projects/openmm>.
- Daylight Theory: SMARTS – A Language for Describing Molecular Patterns (August 2017). <http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html>.
- A. Hjorth Larsen, J. Jørgen Mortensen, J. Blomqvist, I.E. Castelli, R. Christensen, M. Dułak, J. Friis, M.N. Groves, B. Hammer, C. Hargus, E.D. Hermes, P.C. Jennings, P. Bjerre Jensen, J. Kermode, J.R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K.S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K.W. Jacobsen, J. Phys.: Condens. Matter 29 (27), 273002 (2017).
- M.E. Fortunato and C.M. Colina, SoftwareX 6, 7–12 (2017).
- M.E. Fortunato and C.M. Colina, pysimm. <https://github.com/polysimtools/pysimm>.
- L.J. Abbott, K.E. Hart and C.M. Colina, Theor. Chem. Acc. 132 (3), 1334 (2013).
- M. Girard, A. Ehlen, A. Shakya, T. Bereau and M.O. de la Cruz, Comput. Mater. Sci. 167 (May), 25–33 (2019).
- Hoobas Github Repository. <https://bitbucket.org/NUaztec/hoobas/src/master/>.
- Indigox Github Repository. <https://github.com/allison-group/indigox>.
- N.M. O'Boyle, M. Banck, C.A. James, C. Morley, T. Vandermeersch and G.R. Hutchison, J. Cheminform. 3 (10), 1–14 (2011).
- Open Babel Github Repository. <https://github.com/openbabel>.
- D.L. Mobley, C.C. Bannan, A. Rizzi, C.I. Bayly, J.D. Chodera, V.T. Lim, N.M. Lim, K.A. Beauchamp, D.R. Slochower, M.R. Shirts, M.K. Gilson and P.K. Eastman, J. Chem. Theory Comput. 14 (11), 6076–6092 (2018).
- C. Zanette, C.C. Bannan, C.I. Bayly, J. Fass, K. Michael, M.R. Shirts, J.D. Chodera and D.L. Mobley, J. Chem. Theory Comput. 15, 402–423 (2019).
- D.L. Mobley, open-forcefield-group/smirff99Frosst: Version 1.0.1 Zenodo (September 2016). <https://doi.org/10.5281/zenodo.154235>.
- WebFF Github Repository. <https://github.com/usnistgov/WebFF-Documentation>.
- L.O. Hedges, A.S.J.S. Mey, C.A. Laughton, F.L. Gervasio, A.J. Mulholland, C.J. Woods and J. Michel, J. Open Source Softw. 4 (43), 1831 (2019).
- A. Jain, S.P. Ong, W. Chen, B. Medasani, X. Qu, M. Kocher, M. Brafman, G. Petretto, G.M. Rignanese, G. Hautier, D. Gunter and K.A. Persson, Concurr. Comput.: Practice Exp. 27 (17), 5037–5059 (2015), CPE-14-0307.R2.
- FireWorks Github Repository. <https://github.com/materialsproject/fireworks>.
- C.L. Phillips, J.A. Anderson and S.C. Glotzer, J. Comput. Phys.230 (19), 7191–7201 (2011).
- A.Z. Panagiotopoulos, N. Quirke, M. Stapleton and D.J. Tildesley, Mol. Phys. 63 (4), 527–545 (1988).
- A.Z. Panagiotopoulos, Mol. Phys. 61 (4), 813–826 (1987).
- D.J. Adams, Mol. Phys. 29 (1), 307–311 (1975).
- J. Ilja Siepmann and D. Frenkel, Mol. Phys. 75 (1), 59–70 (1992).
- M.G. Martin and J.I. Siepmann, J. Phys. Chem. B 102 (14), 2569–2577 (1998).
- M.S. Shah, J.I. Siepmann and M. Tsapatsis, AIChE J. 63 (11), 5098–5110 (2017).
- Y. Nejahi, M. Soroush Barhaghi, J. Mick, B. Jackman, K. Rushaidat, Y. Li, L. Schwiebert and J. Potoff, SoftwareX 9, 20–27 (2019).
- MoSDeF TraPPE Github Repository. <https://github.com/ahy3nz/mosdef_trappe>.
- C. Fu, Y. Kuang, Z. Huang, X. Wang, Y. Yin, J. Chen and H. Zhou, J. Solid State Electrochem. 15 (11–12), 2581–2585 (2011).
- C. Zhan, C. Lian, Y. Zhang, M.W. Thompson, Y. Xie, J. Wu, P.R. Kent, P.T. Cummings, D. en Jiang and D.J. Wesolowski, Adv. Sci. 4, 1700059 (2017).
- Y. Zhang, B. Dyatkin and P.T. Cummings, J. Phys. Chem. C 123 (20), 12583–12591 (2019).
- J.C. Meyer, A.K. Geim, M.I. Katsnelson, K.S. Novoselov, T.J. Booth and S. Roth, Nature 446 (7131), 60–63 (2007).
- S.M. Mahurin, E. Mamontov, M.W. Thompson, P. Zhang, C.H. Turner, P.T. Cummings and S. Dai, Appl. Phys. Lett. 109 (14), 143111 (2016).
- G. Feng and P.T. Cummings, J. Phys. Chem. Lett. 2 (22), 2859–2864 (2011).
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model. 7 (8), 306–317 (2001).
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A.E. Mark and H.J. Berendsen, J. Comput. Chem. 26 (16), 1701–1718 (2005).
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M.R. Shirts, J.C. Smith, P.M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics 29 (7), 845–854 (2013).
- R.T. McGibbon, K.A. Beauchamp, M.P. Harrigan, C. Klein, J.M. Swails, C.X. Hernández, C.R. Schwantes, L.P. Wang, T.J. Lane and V.S. Pande, Biophys. J. 109 (8), 1528–1532 (2015).
- Graphene-Pore Github Repository. <https://github.com/rmatsum836/Pore-Builder>.
- H.J.C. Berendsen, J.R. Grigera and T.P. Straatsma, J. Phys. Chem. 91 (24), 6269–6271 (1987).
- I.S. Joung and T.E. Cheatham, J. Phys. Chem. B 112 (30), 9020–9041 (2008).
- B. Hess, H. Bekker, H.J.C. Berendsen and J.G.E.M. Fraaije, J. Comput. Chem. 1472, 1463–1472 (1997).
- true_graphene Github Repository. <https://github.com/rmatsum836/true_graphene>.
- true_lipids Github Repository. <https://github.com/uppittu11/true_lipids>.
- A. Weerheim and M. Ponec, Arch. Dermatol. Res. 293 (4), 191–199 (2001).
- K.C. Madison, J. Invest. Dermatol. 121 (2), 231–241 (2003).
- Bilayer Builder Github Repository. <https://github.com/uppittu11/mbuild_bilayer>.
- S. Guo, T.C. Moore, C.R. Iacovella, L.A. Strickland and C. McCabe, J. Chem. Theory Comput. 9 (11), 5116–5126 (2013).
- J.B. Klauda, R.M. Venable, J.A. Freites, J.W. O'Connor, D.J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A.D. MacKerell and R.W. Pastor, J. Phys. Chem. B 114 (23), 7830–7843 (2010).
- H.J.C. Berendsen, J.P.M. Postma, W.F. van Gunsteren, A. DiNola and J.R. Haak, J. Chem. Phys. 81 (8), 3684–3690 (1984).
- W.G. Hoover, Phys. Rev. A 31, 1695–1697 (1985).
- M. Parrinello and A. Rahman, J. Appl. Phys. 52 (12), 7182–7190 (1981).
- S.G. Vilt, Z. Leng, B.D. Booth, C. McCabe and G.K. Jennings, J. Phys. Chem. C 113 (33), 14972–14977 (2009).
- A.Z. Summers, C.R. Iacovella, M.R. Billingsley, S.T. Arnold, P.T. Cummings and C. McCabe, Langmuir 32 (10), 2348–2359 (2016), PMID: 26885941.
- S. Plimpton, J. Comput. Phys. 117 (1), 1–19 (1995).
- T. Darden, D. York and L. Pedersen, J. Chem. Phys. 98 (12), 10089–10092 (1993).
- M. Tuckerman, B.J. Berne and G.J. Martyna, J. Chem. Phys. 97 (3), 1990–2001 (1992).
- J.D. Chodera, J. Chem. Theory Comput. 12 (4), 1799–1805 (2016), PMID: 26771390.
- M.R. Shirts and J.D. Chodera, J. Chem. Phys. 129 (12), 124105 (2008).
- C.S. Adorf, V. Ramasubramani, B.D. Dice, M.M. Henry, P.M. Dodd, and S.C. Glotzer, . glotzerlab/signac Zenodo <https://doi.org/10.5281/zenodo.2581327>
- Tribology Example Github Repository. <https://github.com/daico007/TRUE-nanotribology>.