
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The photochemical interconversion between 1,3-cyclohexadiene (CHD) and all-cis-hexatriene (cZc-HT) is reinvestigated using spin-flip time-dependent density functional theory in combination with various hybrid functionals, BHHLYP functional showing the best performance. The critical geometries of the ground, S, and the first two excited-state, S
and S
, potential energy surfaces, such as, various minima, transition state, minimum-energy crossing points between S
/S
and S
/S
show an excellent agreement with those obtained by multireference wave function methods. Our results show how a low-cost method based on DFT can successfully describe and characterise the most important geometries on the potential energy surfaces along the ring-opening/closure reaction coordinate involved in the CHD to cZc-HT photoconversion.
GRAPHICAL ABSTRACT
1. Introduction
The photochemical interconversion between 1,3-cyclohexadiene (CHD) and all-cis-hexatriene (cZc-HT), that plays a crucial role in the photobiological synthesis of vitamin D [Citation1,Citation2], is one of the most widely studied photochemical electrocyclic reactions [Citation3,Citation4]. The nature of this photoconversion has been the subject of a number of both experimental and theoretical investigations [Citation3–9], and it is often considered as simplified model for the investigation of the photophysical and photochemical properties of important macromolecules, that contain such (4n+2) photo electrocyclic reaction as their central building block. These photoactive molecules are relevant for modern technologies, for example, application of molecular photoswitches [Citation10,Citation11], photomolecular motors [Citation11], nanomachines [Citation12] in photomedicine [Citation13] and molecular electronic devices [Citation14,Citation15].
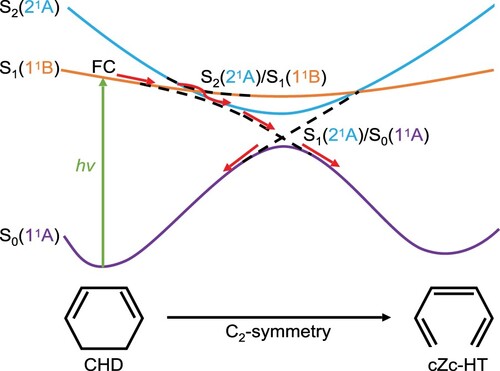
The present status of experimental and theoretical studies on the photochemistry of CHD has been already reviewed by several authors [Citation5,Citation6,Citation9,Citation16–23] and many aspects of the CHD to cZc-HT photochemical interconversion have been clarified. Much of these works have focused on elucidating the ultrafast photoinduced dynamics of CHD through critical points on the potential energy surfaces, such as conical intersections (CIs). Even though it appears relatively simple, this is a very complex subject that led to numerous controversies in the field [Citation9,Citation19,Citation22,Citation24,Citation25] and there exist contradictory experimental and theoretical findings [Citation5,Citation16,Citation17,Citation19,Citation22]. However, it is now widely accepted that CHD to cZc-HT photochemical interconversion occurs via three consecutive steps [Citation16,Citation18,Citation19,Citation21,Citation22] (see Figure ); (i) Photoexcitation of the CHD from the ground state S (belongs to the irreducible representation 1
A under C
symmetry) to the first excited state S
(belongs to the irreducible representation 1
B under C
symmetry) at the Franck-Condon (FC) region. (ii) Rapid decay from the excited state 1
B to the “dark” excited state 2
A. (iii) Decay from 2
A to either the ground state of the cZc-HT or CHD. To the best of our knowledge, the photoreversion of cZc-HT to CHD has not been observed, since cZc-HT structure does not remain in the thermal equilibrium, and subsequently derive into the predominant trans-HT structures via the thermal reactions [Citation6].
Figure 1. Schematic representation of the CHD to cZc-HT photochemical interconversion. The dashed lines represent the crossing between excited states S(2
A) and S
(1
B), and S
(2
A) and the ground state S
(1
A). FC refers to the Franck-Condon region.
The progression of the CHD electrocyclic ring opening described by this scheme is mediated mainly by two CIs, S/S
and S
/S
. Theoretical studies have been performed using multireference wave function methods [Citation16,Citation18,Citation22], density functional theory (DFT) and time-dependent density functional theory (TDDFT) [Citation19,Citation21] in order to locate and characterise these important geometries. The complete active space self-consistent field method (CASSCF) is one of the multireference methods typically used to study the ring-opening/closure photoreaction between CHD and cZc-HT [Citation3,Citation7–9,Citation26]. Usually, the active space employed with CASSCF consists of 6 electrons and 6 orbitals (4
and 2
) for CHD and 6 electrons and 6 orbitals (6
) for cZc-HT [Citation9]. Although this method has satisfactorily described the reaction path between the states 2A and 1A [Citation7,Citation9,Citation26], it fails to reproduce the correct order of the states 1B and 2A in the FC region [Citation5,Citation7–9]. This fact has limited the CASSCF method to describe this reaction process approximately after the crossing between S
and S
[Citation8,Citation9]. Santolini et al. [Citation27] have obtained the correct order of the states in the FC region for the butadiene-like molecules by significantly increasing the size of the restricted active space (RAS). Although this demonstrates that the correct order of the states can be qualitatively achieved, the process of obtaining them for more complicated systems can be hard, and demands an increase in computational complexity [Citation22,Citation27]. The second-order perturbation theory for CASSCF method (CASPT2) is another widely used multireference method that has shown very good results [Citation5,Citation18,Citation22]. Unlike CASSCF, this method computes the correct order of the states in the FC region. Mori and Kato [Citation18] show an interesting comparison between extensions based on CASSCF and CASPT2. They concluded that the dynamic electron correlation is an important factor for the correct description of the energies and geometries [Citation18]. Recently, Poliak et al. [Citation22] used an extended multistate CASPT2, XMS-CASPT2, that offers a good balance of the dynamic electron correlation, for surface hopping simulations and provided a detailed description of the photodeactivation mechanism.
The DFT method, in contrast to multireference methods, has the ability to describe the dynamic electron correlation [Citation28]. A previous work based on this method could capture the right order of states in the FC region [Citation29], which were consistent with the results using CASPT2 [Citation7]. However, DFT, and therefore TDDFT can not describe the nondynamic correlation [Citation28]. Additionally, TDDFT shows serious problems to describe CIs, especially the CIs that involve the S [Citation30,Citation31]. Recently, Filatov et al. [Citation21], used state-interaction, state-averaged spin-restricted ensemble-referenced Kohn-Sham (SSR) method, and were able to provide a correct description of the energies, geometries of the S
, and the CI
. However, the CI
was not described since their studies were confined to the dynamic of CHD ring opening on the S
state [Citation21]. An extension to TDDFT proposed by Shao et al. [Citation31], called spin-flip time-dependent density functional theory (SF-TDDFT), developed to describe diradicals with strong nondynamical correlation, has shown a great performance for describing CIs in ethylene [Citation32], uracil [Citation33] and cis-stilbene [Citation34]. Within this approach, a high-spin triplet state is chosen as the initial reference state allowing that the S
and the single excited states to be treated at the same footing. SF-TDDFT recovers both nondynamical and dynamical correlation from SF and DFT, respectively [Citation31,Citation35]. These features have made SF-TDDFT a great alternative to address photochemical processes. Here, we use SF-TDDFT to study and determine the important geometries at the potential energy profile of CHD to cZc-HT photochemical interconversion, with a special focus on locating and characterising the S
/S
and S
/S
CIs.
This paper is organised as follows: Section 2 briefly describes the underlying theoretical methods used in this work. Computational details are summerised in Section 3. Our results and discussions are presented in Section 4 and finally our concluding remarks are given in Section 5.
2. Theoretical methods
2.1. Orbital and state correlation diagrams
The CHD to cZc-HT photochemical interconversion can be depicted qualitatively using the conservation of orbital symmetry, i.e. following the Woodward-Hoffmann rules [Citation36] for a pericyclic reaction. Similar to the description made for the ring closure of 1,3-butadiene in reference [Citation37] using the Woodward-Hoffmann rules, we shall illustrate the photochemical interconversion between CHD and cZc-HT. Figure describes an orbital correlation diagram when the absorption of a photon by the CHD leads to the excitation of an electron from the bonding orbital 2π to the antibonding orbital 3π. The conrotatory (4n+2 electron in π system) path of the excited CHD leads to the excited cZc-HT keeping the same orbital symmetry [Citation37,Citation38]. However, the suggestion of an intermediate excited state cZc-HT generates a problem since it is known that CHD, after photoexcitation, return to either its ground state or to the ground state of cZc-HT, i.e. without the formation of an intermediate excited state cZc-HT [Citation4]. This discrepancy can be resolved if one considers the overall states of CHD and cZc-HT via state correlation diagram presented in Figure . For doing so, we used the direct product between the symmetry specie of the individual occupied orbitals satisfying the following rules: S × S = S; S × A = A and A × A = S, where S (with character 1) means symmetric with respect to a symmetry element and A (with character -1) means antisymmetric with respect to a symmetry element. All doubly occupied orbitals are always totally symmetric. Because the conrotatory path preserves the rotation around the C axis, we can classify the relevant states in terms of S and A with respect to this axis [Citation37,Citation38]. Since CHD (1σ,1π,2π) is correlated with cZc-HT (1π,2π,4π) (see Figure ), it follows that the CHD (1
1
2
,
S) is correlated with cZc-HT (1
2
4
,
S), where
S denotes the result of the direct product between the symmetry S × S = S and the superscript 1 means that the electrons are in a singlet configuration (see Figure ).
Figure 2. Orbital correlation diagram for the photochemical interconversion of the CHD to cZc-HT. The orbitals of CHD and cZc-HT are ordered in energy and labelled as symmetric (S) and antisymmetric (A) under C symmetry. Dashed lines relate empty orbitals with the same symmetry and solid lines relate occupied orbitals with the same symmetry.
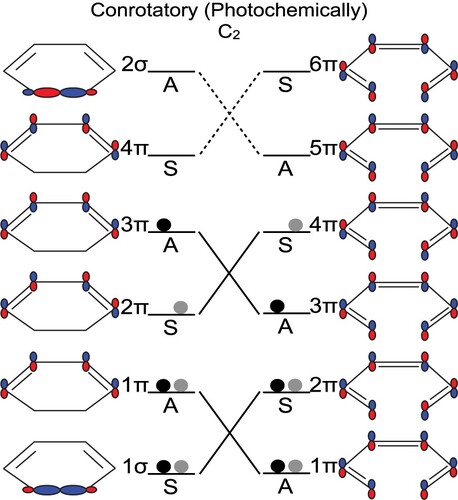
Figure 3. Scheme of the state correlation diagram for the photocyclisation of CHD to cZc-HT. Dashed lines connect molecular orbitals with the same symmetry and same multiplicity, and solid blue lines connect molecular orbitals satisfying the non-crossing rule.
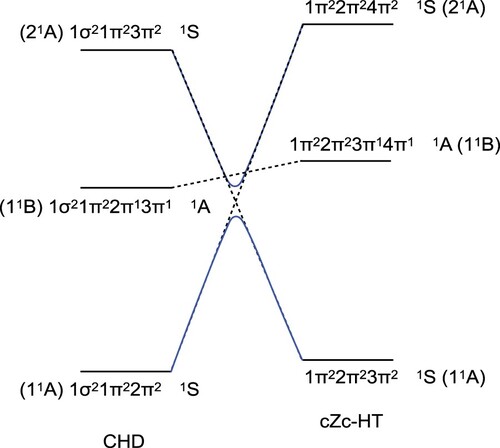
Following the same protocol in reference [Citation37] for the ring closure of 1,3-butadiene example, the dashed lines in Figure stand for the connections between the states with the same symmetry and multiplicity. Note that it is not allowed to have crossings between the energy levels with the same symmetry (non-crossing rule) [Citation37,Citation39,Citation40], therefore the blue lines are used to remark what happens. However, the non-crossing rule is only valid for diatomic molecules. Thus, in polyatomic molecules, this rule loses its validity and two electronic states with the same symmetry and multiplicity may cross at a conical intersection [Citation26,Citation41,Citation42]. Therefore, the dashed lines show the behaviour expected.
In the literature [Citation3,Citation17], the molecular orbitals of CHD and cZc-HT are frequently represented with the Mulliken symbols for irreducible representations of the C point group. Thus, the molecular orbitals 1
1
2
, 1
1
2
3
and 1
1
3
of CHD are represented as 1
A, 1
B, and 2
A, respectively and in the same way, the molecular orbitals 1
2
3
, 1
2
3
4
and 1
2
4
of cZc-HT are represented as 1
A, 1
B, and 2
A, respectively. Now lets consider the following case, incoming photon generates population to the first excited state (1
B) of CHD, then one we can think of a population transfer to the first excited state of cZc-HT (1
B), as it was shown in the analysis of the individual orbital in Figure , but as mentioned above, this behaviour has not been observed. An alternative doorway can be opened if there is a crossing (i.e. internal conversion driven by the nonadiabatic interaction) between the bright 1
B and the dark 2
A states of CHD, through which the population of the bright state is transferred to the dark state. The inclusion of the dark 2
A state, proposed by van der Lugt and Oosterhoff [Citation24], allows an internal conversion between the blue line and the dashed line (see Figure ). Subsequently, a second internal conversion occurs from 2
A to 1
A, that results in the population transfer to the ground state of cZc-HT or CHD without the need to pass through the first excited state of cZc-HT, 1
B.
2.2. Spin-Flip TDDFT
The main application of TDDFT is the calculation of excited states which can be accessed through linear-response (LR) theory. In LR-TDDFT [Citation43], the TD perturbation must be small enough such that the perturbation does not change completely the ground state of the system. Within LR-TDDFT, the non-Hermitian eigenvalue equation
(1)
(1) is solved for obtaining the excitation energies Ω and transition amplitudes X and Y. Here, the coupling matrices A and B are:
(2)
(2) and
(3)
(3) in which
(4)
(4) and
(5)
(5) where indexes
and
refer to occupied and unoccupied spin-orbitals, respectively, and
is the fraction of Hartree-Fock (HF) exchange in the exchange-correlation (xc) functional
. The latter is the xc kernel which is given by the functional derivative of the xc energy. In SF-TDDFT method, that was introduced by Shao et al. [Citation31] in 2003, a high-spin (
) triplet state, with two unpaired α-electron, is chosen as a reference state. Note that for conventional LR-TDDFT calculations, only
and
blocks are considered. In SF-TDDFT, the target states must be
, thus only
blocks are taken into account. Using a collinear exchange-correlation functional kernel, i.e.
, and considering the orthogonality between the occupied α and β spin-orbitals, the coupling matrices equations, Equation Equation2
(2)
(2) and Equation Equation3
(3)
(3) , reduce to the following expressions
(6)
(6) and
(7)
(7) Note that Equation Equation7
(7)
(7) is a result of using a collinear kernel which is similar to set B = 0 within the Tamm-Dancoff approximation (TDA)[Citation44] in the LR-TDDFT. Thus, the SF-TDDFT eigenvalue equation to be solved is given by
(8)
(8) SF-TDDFT is a very efficient method for describing excitation energies, topology around conical intersections, in particular those involving ground state, and excited-state reaction pathways. However, this method has one big drawback that is spin contamination and an effective state-tracking algorithm is required whenever SF-TDDFT is used. A straightforward way to do this, is to monitor the change in the excited-state transition density. An example, used in this work, is the state-tracking algorithm that is based on the overlap of the attachment/detachment densities at successive steps that can avoid any problems that may be introduced by sign changes in the orbitals or configuration-interaction coefficients. It has been shown by Herbert et al. [Citation45] that although a state-tracking algorithm, in principle improve the performance of SF-TDDFT for CIs or minimum energy crossing points (MECP) optimisation, the spin adopted (SA)SF-TDDFT, being free of spin contamination and just as computationally efficient as SF-TDDFT, is a safer remedy.
2.3. Minimum-Energy crossing point optimisation
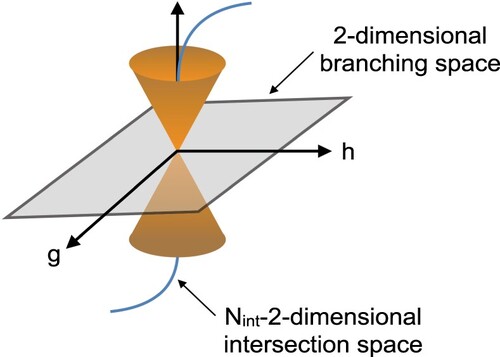
In nonadiabatic process, the crossing between two electronic states (I and J) along a -2-dimensional seam (
is the internal degrees of freedom) has a special significance [Citation46,Citation47] since the MECPs along this seam are one of the factors controlling the excited state nonadiabatic deactivation pathways [Citation46,Citation48]. The degeneracy between two states I and J is called conical intersection (CI) because the form of the two PESs around of seam has a conical shape, that arises because the degeneracy between the two states, I and J, is lifted along two orthogonal vectors; the difference gradient (DG) vector
(9)
(9) which points in the direction of maximal energy splitting and the nonadiabatic coupling (NAC) vector
(10)
(10) which points in the direction of maximal nonadiabatic interaction (see Figure ). These two vectors describe the 2-dimensional branching space defining the CI. For any electronic structure method that has analytic excited-state gradients, the
vector is already available, while computing
vector is not an easy task, since it requires first-derivative of the electronic wave function with respect to the nuclear coordinates. Although NAC is needed for optimisation of MECPs, there exist efficient MECPs optimisation algorithms that do not necessarily need calculation of NAC vector [Citation49–51]. Recently, analytic derivative couplings for SF-TDDFT has been reported [Citation52], thus here we rely on an optimisation algorithm that make use of both
and
vectors, i.e. the projected-gradient algorithm of Bearpark et al. [Citation49] which is implemented in Q-CHEM [Citation53]. This algorithm consists of minimising along the following gradient:
(11)
(11) where
(12)
(12) is the normalised DG vector,
(13)
(13) is the mean energy gradient for states I and J, and
(14)
(14) is a projector operator onto the seam space where
(15)
(15) It has been shown that the projected gradient algorithm, that uses both
and
, reduces the number of iterations necessary to converge the MECP [Citation52,Citation54]. This fact makes the projected gradient algorithm more efficient than those algorithms that only use the vector
.
Figure 4. Schematic representation of a CI between two PESs in the -2-dimensional space. Two orthogonal vectors define the 2-dimesional branching space: g which points in the direction of maximal energy splitting and h which points in the direction of maximal nonadiabatic interaction.
3. Computational details
Ground state geometry optimisation of CHD and cZc-HT were performed at ωB97X-D/cc-pVDZ level of theory [Citation55,Citation56], including Grimme's dispersion correction [Citation57]. Subsequent frequency calculations were undertaken to confirm the nature of the minima. The vertical excitation energies of the first (S) and the second (S
) excited state of CHD and cZc-HT and their optimised geometries were obtained using SF-TDDFT/cc-pVDZ in combination with the ωB97X-D, B5050LYP, and BHHLYP xc functionals, for bench-marking purpose, from which BHHLYP was finally selected. Relaxed potential energy surface (PES) scans of S
, S
and S
were carried out, along C
–C
bond distance, at SF-TDDFT(BHHLYP)/cc-pVDZ level of theory. The geometries of all identified minima and transition state structure have been individually optimised. Furthermore, the nature of the transition state has been confirmed using the intrinsic reaction coordinate (IRC) algorithm, which is essentially a series of steepest descent steps going downhill from the transition state to the adjacent educt and product states. The MECPs between S
/S
and S
/S
were located using SF-TDDFT(BHHLYP)/cc-pVDZ. Finally, natural transitions orbitals (NTOs) of important geometries were computed using SF-TDDFT(BHHLYP)/cc-pVDZ. All the calculations were performed using Q-Chem 5.1.2 package [Citation53]. Cartesian coordinates of all the relevant structures are given in the Supporting Information (SI).
4. Results and discussions
4.1. Stationary geometries
Ground state optimised geometries of CHD and cZc-HT molecules are illustrated in Figures (a) and (b), respectively. The distance between C and C
(reaction coordinate here) is 1.53 Å for the closed (CHD) and 3.44 Å for the open (cZc-HT) ring. Both geometries of CHD and cZc-HT show C
symmetry in their S
minimum. C
has two irreducible representations, A (properties are symmetric to both the identity operation E and a rotation 180 degree rotation around C
axis) and B (properties are symmetric with respect to the identity operation E). The ground state optimised geometries are in good agreement with previous works [Citation21,Citation22].
Figure 5. Optimised geometries with DFT(ωB97X-D)/cc-pVDZ: (a) ground state CHD, (b) ground state cZc-HT. Optimised geometries with SF-TDDFT(BHHLYP)/cc-pVDZ: (c) CI (from CHD), (d) CI
, (e) CI
(from cZc-HT), (f) S
, (g) S
and (h) S
transition state. Distances are in Å.
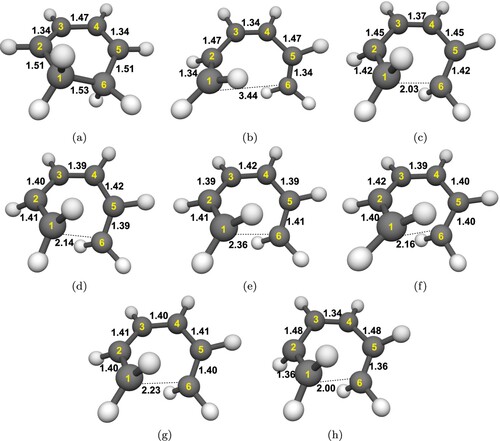
The S geometry optimisations of CHD and cZc-HT molecules both converged to the same local minimum with an energy of 3.92 eV, symmetry of C
, and the C
–C
distance of being 2.16 Å (see Figure (f)). Please note that the C
–C
bond length is elongated (by 0.63 Å) and shortened (by 1.28 Å) compared with CHD-S
and cZc-HT-S
, respectively. Such structural changes make it evident that vibrational relaxation on S
leads to the ring opening of the CHD. The energy of S
is in a reasonable agreement with previously reported MS-CASPT2 results (4.40 eV), with the C
–C
distance being larger, ≈ 0.5 Å compared with the corresponding distance using MS-CASPT2 [Citation18], and in both methodologies
vibrational relaxation leads to the opened ring structure. Similarly, the S
geometry optimisations of the CHD and cZc-HT molecules also converged to the same local minimum with an energy of 4.34 eV, symmetry of C
, and the C
–C
distance of being 2.23 Å (see Figure (g)). It must be noted that although vibrational relaxation on the S
leads to the ring opening, but unlike S
, the S
has C
symmetry similar to CHD-S
and cZc-HT-S
. To the best of our knowledge, no theoretical and experimental results have been reported for the S
. It must be noted that unconstrained geometry optimisation of the S
state leads directly to the crossing region between the S
and S
states, while unconstrained geometry optimisation of the S
state leads to the crossing region between S
and S
states. Both of these points will be discussed later. Our results shows that interconversion between CHD and cZc-HT in the S
proceed via a transition state with C
symmetry, and the C
–C
distance of being 2.0 Å (see Figure (h)). The energy barrier for this process amounts to 3.4 eV, indicating that thermal electrocyclic reaction can be excluded from being relevant in the CHD to cZc-HT interconversion.
4.2. Vertical excitation energies
Table summarises the vertical excitation energies (VEE) for the two lowest singlet excited states of the CHD and cZc-HT molecules calculated with TDDFT (various functionals) as well as experimental and previous theoretical studies using multi reference wave function-based methods. Recently, Polyak et al. [Citation22] have performed a comprehensive theoretical study on the CHD photodissociation using the XMS-CASPT2 method. They reported 5.13 eV for the S VEE and 6.28 eV for the S
VEE, that are very close to the experimental results [Citation5] (see Table ). Comparing the performance of SF-TDDFT with wave function-based methods listed in Table , it is apparent that functionals containing larger amount of HF exchange (>50%), perform in a closer agreement (difference of
eV for S
and
eV for S
) with the corresponding value obtained at XMS-CASPT2/cc-pVDZ level of theory. For all level of theories, the order of states remains essentially the same; the S
state with a 1
B symmetry being the bright
state and the S
with the 2
A symmetry being the dark
state.
Table 1. Vertical excitation energies in eV for the two lowest singlet excited states of CHD and cZc-HT.
Additionally, for the sake of completeness, we calculated the VEE of cZc-HT (see Table ). Similar to the CHD molecule, all functionals shown that the S state with a 1
B symmetry is the bright
state and the S
with the 2
A symmetry is the dark
state (The state-averaged NTO involved in the transitions are shown in Supporting Information Figure 1). To the best of our knowledge, no experimental results is available for the cZc-HT molecule. We observed almost no difference in the excitation energies obtained using BHHLYP and B5050LYP. Earlier studies [Citation31–33] have revealed that BHHLYP is a suitable functional when describing excited sate and MECP optimisations within SF-TDDF methodology. Follwoing these conclusions, we will restrict ourselves to BHHLYP functional for further discussion.
4.3. Minimum energy crossing points
As well known [Citation46–48], the excited state nonadiabatic dynamics is essentially controlled by the energies of the minima of the various surfaces, the various conical intersection seams, and MECPs along these seams. In this section, we discuss two MECPs that are known to play significant roles in actual photochemical interconversion of CHD to cZc-HT, namely, the CIs between S and S
and S
and S
, denoted as CI
and CI
, respectively. Optimised structures of these geometries are shown in Figure and their relatives energies to S
of CHD are listed in Table . Furthermore, the energies of the two states involved in the MECP optimisation, norm of derivative coupling (i.e. NACs) and the C
–C
distance along the MECPs optimisation trajectories, are plotted in Figures – .
Figure 6. (a) CI: potential energies curves (i), norm of derivative coupling (ii) and distance between the reaction carbons C
–C
(iii) along the S
/S
MECP optimisation trajectory of the CHD. (b) CI
: potential energies curves (i), norm of derivative coupling (ii) and distance between the reaction carbons C
–C
(iii) along the S
/S
MECP optimisation trajectory of the CHD. The energies are relatives to the S
energy of the CHD.
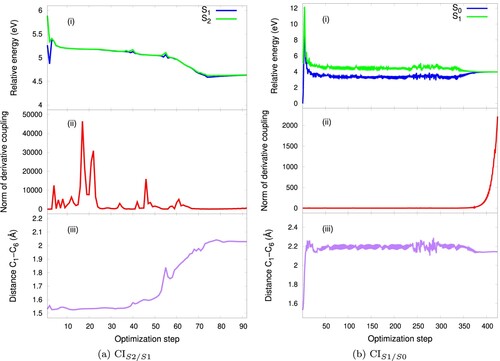
Figure 7. (a) CI: potential energies curves (i), norm of derivative coupling (ii) and distance between the reaction carbons C
–C
(iii) along the S
/S
MECP optimisation trajectory of the cZc-HT. (b) CI
: potential energies curves (i), norm of derivative coupling (ii) and distance between the reaction carbons C
–C
(iii) along the S
/S
MECP optimisation trajectory of the cZc-HT. The energies are relatives to the S
energy of the CHD.
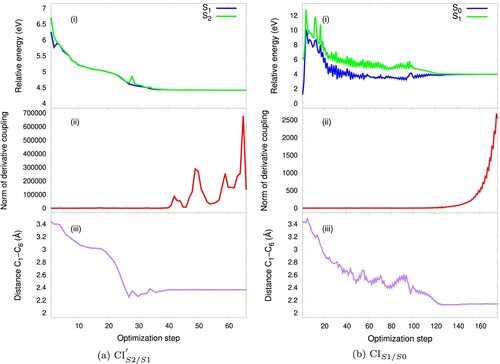
Table 2. C –C bond distances and energies at different level of theories. The distances are in Å and after the slash, the energies relative to S energy of CHD are in eV.
–C bond distances and energies at different level of theories. The distances are in Å and after the slash, the energies relative to S energy of CHD are in eV.
CI. The CHD and cZc-HT S
geometries served as the starting point in both calculations searching for the MECPs along the crossing seam between the S
and S
states. Starting from CHD, the optimisations converged to a minimum, CI
, with an energy of 4.63 eV, symmetry of C
, and the C
–C
distance of being 2.03 Å (see Table and Figure (c)). Interestingly, the nature of this structure is very similar to the S
with respect to the symmetry, energy (difference of 0.29 eV higher) as well as the C
–C
distance (difference of 0.2 Å). The latter will indicate that the CI
is located in the close vicinity of the S
. The geometry of this MECP is in good agreement with previous works [Citation18,Citation22]. The energy of the CI
is 0.63 eV lower than the energy of the CHD molecule at the FC point, according to the SF-BHHLYP/cc-pVDZ method; this energy difference is 0.74 eV for XMS-CASPT2/cc-pVDZ and 0.76 eV for MS-CASPT2 calculations (see Table ). Thus, it is natural to assume that, for all these three methods, the CHD will undergo vibrational relaxation downhill from the FC region and reach the CI
. Starting from cZc-HT, the optimisations converged to a minimum, CI
, with an energy of 4.42 eV, symmetry of C
, and the C
–C
distance of being 2.36 Å (see Table and Figure (e)). Interestingly, the nature of this structure is also very similar to the S
with respect to the symmetry, energy (difference of 0.08 eV) as well as the C
–C
distance (difference of 0.13 Å). We observe that the CI
is much closer to the S
. The energy of the CI
is 1.81 eV lower than the energy of the cZc-HT molecule at the FC point, according to the SF-BHHLYP/cc-pVDZ method (see Table ). Note that the CI
is only 0.21 eV lower than the CI
, and the S
is located between these two CIs, being closer to the former (see Table ). To the best of our knowledge, the CI
has not been previously reported. In sum, we propose that after photoexcitation to S
, both the CHD and cZc-HT undergo vibrational relaxation to S
after passing through low-lying MECPs between S
and S
, i.e. CI
and CI
, respectively. This will be discussed later in the context of the PESs for the overall the CHD to cZc-HT photochemical interconversion (see Figure ).
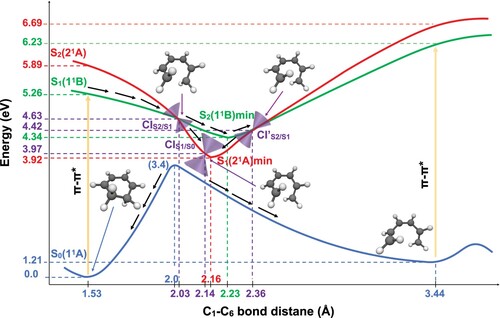
Figure 8. Schematic representation of the PESs of CHD/cZc-HT photochemical interconversion. The reaction coordinate is the C–C
bond distance. The 1
A (ground state) is in blue, the state 1
B is in green and the state 2
A is in red. The curves 1
B and 2
A are a pictorial description that connects the important geometries computed with SF-BHHLYP/cc-pVDZ. The curve 1
A is a PES scan trough the C
–C
bond distance computed with SF-BHHLYP/cc-pVDZ. The cones in purple represent the MEPCs. The arrows depict how the photochemical interconversion process follow after an absorption of a photon generates the population of the first excited state (1
B) of CHD.
CI. The CHD and cZc-HT S
geometries served as the starting point in both calculations searching for the MECPs along the crossing seam between the S
and S
states. Interestingly, the optimisations converged, independent of starting from CHD or cZc-HT, to the same local minimum with an energy of 3.97 eV, symmetry of C
, and C
–C
distance of being 2.14 Å (see Table and Figures (d)). Interestingly, the nature of this structure is very similar (almost identical) to the S
with respect to the symmetry, energy (difference of 0.05 eV higher) as well as the C
–C
distance (difference of 0.02 Å). The latter speaks in favour of the fact that the CI
is located extremely close to the S
. The geometry of this MECP is in good agreement with previous works [Citation18,Citation21,Citation22]. The energy of the CI
is 0.66 eV lower than the energy of the CI
geometry, according to the SF-BHHLYP/cc-pVDZ method; the corresponding energy difference is energy is 0.59 eV for XMS-CASPT2/cc-pVDZ and 0.87 eV for MS-CASPT2 calculations (see Table ). Thus, it is natural to assume that, for all these three methods, the CHD will go downhill from the CI
towards CI
, and from there towards either to the S
, that is slightly lower in energy (≈0.05 eV), or to the S
surface. This fact suggests that the relaxation process from CI
to CI
is accelerated, supporting the hypothesis formulated by Garavelli et al. [Citation7] about the ultrashort relaxation lifetime reported after S
/S
crossing [Citation6], i.e. there is a higher probability of hitting a CI during motion along the bottom of S
. The similar scenario holds if one considers relaxation starting from CI
.
4.4. Potential energy surface
In this section, the PESs for the lowest three singlet states, S, S
and S
, along C
–C
coordinate, are investigated in more detail, shedding further light on the CHD/cZc-HT photochemical interconversion. We use the S
, S
, S
to indicate adiabatic states and the
,
and
to refer diabatic states. As it is depicted in Figure , the the S
PES has two minima corresponding to i) the CHD with C
–C
distance of 1.53 Å (closed ring) and ii) the cZc-HT with C
–C
distance of 3.44 Å (open ring). The energy barrier for connecting these two minima, along C
–C
coordinate, is 3.4 eV (78.41 kcal/mol), via the transition state structure at 2.0 Å. This large barrier practically excludes the thermal electrocyclic interconversion as a plausible reaction, and supports the photochemical electrocyclic reaction being the operative pathway.
After absorption of a photon, the CHD is vertically excited, from its S, to the first (bright) singlet excited state, S
(1
B), with an energy of 5.26 eV. From the FC region, the CHD undergoes a vibrational relaxation, during which it hits the CI
(at 2.03 Å and energy of 4.63 eV), via which internal conversion occurs. This behaviour is typical when the CI is accessible from the FC region without significant energy barriers that is the case here. From this critical point, the reaction pathway may bifurcate in two directions, depending on which character the wave function takes after the system leaves the crossing region. Two scenarios can be defined; 1) if the diabatic character changes from 1
B to 2
A, then the system will evolve directly toward the CI
(at 2.14 Å with an energy of 3.97 eV) that triggers an ultra-fast internal conversion process and provides a funnel of fast access to the ground state, on which the system can evolve either to the S
of CHD or the S
of cZc-HT. 2) if the diabatic character does not change, system evolves towards the S
(1
B), from where the low-lying CI
(at 2.36 Å and energy of 4.42 eV) is accessible without significant energy barriers (only 0.08 eV higher), keeping in mind the excess vibrational energy after photoexcitation. After internal conversion through the CI
, the system will undergo a vibrational relaxation through which it slides down again hitting the CI
, that acts as again as a doorway for an ultrafast internal conversion to the ground state, on which the system can again evolve either to the S
of CHD or the S
of cZc-HT. To the best of our knowledge, the excited state deactivation pathway via the CI
, has not been reported so far.
5. Conclusions
In the present work, we have applied SF-TDDFT to reinvestigate the photochemical interconversion between CHD and cZc-HT. Our results reveal that the SF-TDDFT/cc-pVDZ in combination with the BHHLYP functional was able to describe successfully the critical geometries of the ground, S, and the first two excited-state, S
and S
, potential energy surfaces, such as, various minima, transition state, minimum-energy crossing points between S
/S
and S
/S
, being in good agreements with the corresponding structures obtained by multireference wave function methods [Citation16,Citation18,Citation22] and a variant of DFT [Citation21]. Additionally we have identified an additional MECP between S
and S
, namely CI
, that opens an alternative deactivation channel for the excited CHD molecule, and subsequently fully described the photochemical interconversion between CHD and cZc-HT. This suggest that SF-TDDFT could be a good low-cost method to study complex molecules that contain the CHD chromophore as their central backbone, such as dithienylethene molecular photo switches relevant in molecular electronic devices [Citation11,Citation58].
Supplemental Material
Download PDF (793.4 KB)Acknowledgments
This work is part of Innovational Research Incentives Scheme Vidi 2017 with project number 016.Vidi.189.044, which is financed by the Dutch Research Council (NWO).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- E. Havinga and J. Schlatmann, Tetrahedron 16 (1), 146–152 (1961).
- N.A. Anderson, J.J. Shiang, and R.J. Sension, J. Phys. Chem. A 103 (50), 10730–10736 (1999).
- S. Deb and P.M. Weber, Annu. Rev. Phys. Chem. 62 (1), 19–39 (2011). PMID: 21054174.
- B.C. Arruda and R.J. Sension, Phys. Chem. Chem. Phys. 16, 4439–4455 (2014).
- M. Merchán, L. Serrano-Andrés, L.S. Slater, B.O. Roos, R. McDiarmid, and Xing, J. Phys. Chem. A 103 (28), 5468–5476 (1999).
- W. Fuß, W.E. Schmid, and S.A. Trushin, J. Chem. Phys. 112 (19), 8347–8362 (2000).
- M. Garavelli, C.S. Page, P. Celani, M. Olivucci, W.E. Schmid, S.A. Trushin, and W. Fuss, J. Phys. Chem. A 105 (18), 4458–4469 (2001).
- M. Boggio-Pasqua, M.J. Bearpark, M. Klene, and M.A. Robb, J. Chem. Phys. 120 (17), 7849–7860 (2004).
- A. Nenov, P. Kölle, M.A. Robb, and R. de Vivie-Riedle, J. Org. Chem. 75 (1), 123–129 (2010). PMID: 19954144.
- A. Staykov, J. Areephong, W.R. Browne, B.L. Feringa, and K. Yoshizawa, ACS. Nano. 5 (2), 1165–1178 (2011). PMID: 21204563.
- M. Irie, T. Fukaminato, K. Matsuda, and S. Kobatake, Chem. Rev. 114 (24), 12174–12277 (2014). PMID: 25514509.
- J.T. Foy, Q. Li, A. Goujon, J.R. Colard-Itté, G. Fuks, E. Moulin, O. Schiffmann, D. Dattler, D.P. Funeriu, and N. Giuseppone, Nat. Nanotechnol 12, 540 EP– 545 (2017).
- H. Lim, Clinical Photomedicine, (2018)
- S.E. Lyshevski, Nano and molecular electronics handbook, (2016)
- M. Baroncini, M. Canton, L. Casimiro, S. Corra, J. Groppi, M. La Rosa, S. Silvi, and A. Credi, Eur. J. Inorg. Chem. 2018 (42), 4589–4603 (2018).
- H. Tamura, S. Nanbu, H. Nakamura, and T. Ishida, Chem. Phys. Lett. 401 (4), 487–491 (2005).
- H. Tamura, S. Nanbu, T. Ishida, and H. Nakamura, J. Chem. Phys. 124 (8), 084313 (2006).
- T. Mori and S. Kato, Chem. Phys. Lett. 476 (1), 97–100 (2009).
- O. Schalk, T. Geng, T. Thompson, N. Baluyot, R.D. Thomas, E. Tapavicza, and T. Hansson, J. Phys. Chem. A 120 (15), 2320–2329 (2016). PMID: 27018427.
- A.R. Attar, A. Bhattacherjee, C.D. Pemmaraju, K. Schnorr, K.D. Closser, D. Prendergast, and S.R. Leone, Science 356 (6333), 54–59 (2017).
- M. Filatov, S.K. Min, and K.S. Kim, Mol. Phys. 117 (9–12), 1128–1141 (2019).
- I. Polyak, L. Hutton, R. Crespo-Otero, M. Barbatti, and P.J. Knowles, J. Chem. Theory Comput. 15 (7), 3929–3940 (2019). PMID: 31244132.
- T.J.A. Wolf, D.M. Sanchez, J. Yang, R.M. Parrish, J.P.F. Nunes, M. Centurion, R. Coffee, J.P. Cryan, M. Gühr, K. Hegazy, A. Kirrander, R.K. Li, J. Ruddock, X. Shen, T. Vecchione, S.P. Weathersby, P.M. Weber, K. Wilkin, H. Yong, Q. Zheng, X.J. Wang, M.P. Minitti, and T.J. Martínez, Nat. Chem. 11 (6), 504–509 (2019).
- W.T.A.M. Van der Lugt and L.J. Oosterhoff, J. Am. Chem. Soc. 91 (22), 6042–6049 (1969).
- M. Garavelli, P. Celani, M. Fato, M.J. Bearpark, B.R. Smith, M. Olivucci, and M.A. Robb, J. Phys. Chem. A 101 (11), 2023–2032 (1997).
- P. Celani, S. Ottani, M. Olivucci, F. Bernardi, and M.A. Robb, J. Am. Chem. Soc. 116 (22), 10141–10151 (1994).
- V. Santolini, J.P. Malhado, M.A. Robb, M. Garavelli, and M.J. Bearpark, Mol. Phys. 113 (13–14), 1978–1990 (2015).
- D. Cremer, Mol. Phys. 99 (23), 1899–1940 (2001).
- E. Tapavicza, A.M. Meyer, and F. Furche, Phys. Chem. Chem. Phys. 13, 20986–20998 (2011).
- J. Gräfenstein and D. Cremer, Phys. Chem. Chem. Phys. 2, 2091–2103 (2000).
- Y. Shao, M. Head-Gordon, and A.I. Krylov, J. Chem. Phys. 118 (11), 4807–4818 (2003).
- N. Minezawa and M.S. Gordon, J. Phys. Chem. A 113 (46), 12749–12753 (2009). PMID: 19905013.
- X. Zhang and J.M. Herbert, J. Phys. Chem. B 118 (28), 7806–7817 (2014). PMID: 24428679.
- Y. Harabuchi, K. Keipert, F. Zahariev, T. Taketsugu, and M.S. Gordon, J. Phys. Chem. A 118 (51), 11987–11998 (2014). PMID: 25181251.
- D. Casanova and A.I. Krylov, Phys. Chem. Chem. Phys. 22, 4326–4342 (2020).
- R.B. Woodward and R. Hoffmann, Angew. Chem. Int. Ed. Engl. 8 (11), 781–853 (1969).
- P. Atkins and R. Friedman, Molecular Quantum Mechanics, (2011).
- E. Anslyn, D. Dougherty, E. Dougherty, and U.S. Books, Modern Physical Organic Chemistry (2006).
- C. Coulson, Valence, (1961).
- K. Yates, in Hückel Molecular Orbital Theory, edited by K. Yates (1978), pp. 239–290.
- H. Köuppel, W. Domcke, and L.S. Cederbaum, in Adv. Chem. Phys. 59–246 (1984).
- M.A. Robb, F. Bernardi, and M. Olivucci, Pure Appl. Chem. 67 (5), 783–789 (1995).
- M.E. Casida, in Recent Advances in Density Functional Methods (1995), Chap. 5, pp. 155–192.
- I. Tamm, (Moscow), (1945), Vol. 9, pp. 449..
- X. Zhang and J.M. Herbert, J. Chem. Phys. 143 (23), 234107 (2015).
- D.R. Yarkony, Rev. Mod. Phys. 68, 985–1013 (1996).
- T.J. Martínez, Nature 467 (7314), 412–413 (2010).
- S. Faraji, S. Matsika, and A.I. Krylov, J. Chem. Phys. 148 (4), 044103 (2018).
- M.J. Bearpark, M.A. Robb, and H.B. Schlegel, Chem. Phys. Lett. 223 (3), 269–274 (1994).
- B.G. Levine, J.D. Coe, and T.J. Martínez, J. Phys. Chem. B 112 (2), 405–413 (2008). PMID: 18081339.
- S. Maeda, K. Ohno, and K. Morokuma, J. Chem. Theory Comput. 6 (5), 1538–1545 (2010). PMID: 26615689.
- X. Zhang and J.M. Herbert, J. Chem. Phys. 141 (6), 064104 (2014).
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H.P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O'Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K.L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill, and M. Head-Gordon, Mol. Phys 113, 184–215 (2015).
- J.M. Herbert, X. Zhang, A.F. Morrison, and J. Liu, Acc. Chem. Res 49 (5), 931–941 (2016). PMID: 27100899.
- J.D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
- T.H. Dunning, J. Chem. Phys. 90 (2), 1007–1023 (1989).
- S. Grimme, J. Comput. Chem. 27 (15), 1787–1799 (2006).
- D. Roke, C. Stuckhardt, W. Danowski, S.J. Wezenberg, and B.L. Feringa, Angew. Chem. Int. Ed. 57 (33), 10515–10519 (2018).