
Abstract
Carbenes are challenging molecular species for quantum chemistry because of the energetic proximity of their singlet and triplet spin states and the sensitive dependence of spin-state energetics on the geometry of the carbene site. Here we use an extended set of aryl-carbenes to evaluate the performance of density functional theory (DFT) approximations as well as of wave function based perturbation theory approaches (orbital-optimised perturbation theory methods OO-MP2 and OO-SCS-MP2) against reference coupled cluster calculations with singles, doubles and perturbative triples conducted with the aid of the domain-based local pair natural orbitals approach, DLPNO-CCSD(T). In addition to the expected functional dependence, our results document a remarkable discordance in the performance of DFT methods in the sense that the functionals that yield the best geometries do not coincide with those that provide the best spin-state energetics. Analysis of the results allows us to propose a series of methods that are expected to perform reliably within certain confidence limits for the title systems. Additionally, methodological issues regarding the reference singlet–triplet gaps obtained by the DLPNO-CCSD(T) approach are discussed.
GRAPHICAL ABSTRACT
1. Introduction
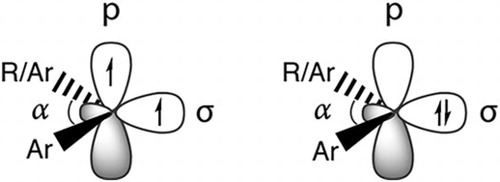
Carbenes are highly reactive organic molecules where a neutral carbon atom has two electrons less than an octet structure [Citation1,Citation2]. In the language of valence bond theory, carbene centres adopt sp2 hybridisation and hence are bent. Their electronic structure and spin state multiplicity can be described in a simplified manner by assuming two electrons to be distributed in two nearly degenerate p and sp2 orbitals (Figure ). Formation of parallel spins in this configuration results in a high-spin triplet state (S = 1), while pairing of the orbitals in the sp2 orbital leads to a singlet state (S = 0). The triplet and singlet carbenes are significantly different species both structurally and electronically. Most carbenes adopt a triplet ground state, but singlet ground state carbenes can also form, or spin-state equilibria established, through interaction with proper substituents and steric restrictions [Citation3–6]. The significant difference in electron distribution between the two spin states results in distinctly different chemical properties and reactivity. For instance, the often higher-lying singlet state participates in reactions rather than the lower-lying triplet state. Therefore, knowledge of accurate singlet–triplet energy gaps are of high importance in carbene chemistry.
Figure 1. Schematic distribution of electrons in the triplet (left) and singlet (right) spin states of the simplest carbene, methylene.
In recent studies, it was shown that localised coupled cluster methods, specifically the domain-based pair natural orbital (DLPNO) coupled-cluster theory [Citation7–9], can be used to assess the energetics of systems as large as a small protein and even larger. This approach can therefore bring the accuracy of the ‘gold standard’ CCSD(T) calculations to much larger systems than those accessible with conventional CCSD(T) calculations. Carbenes are one of the chemically interesting systems where canonical CCSD(T) calculations are absolutely required [Citation10] for achieving chemical accuracy in singlet–triplet gaps (ca. 0.4 kcal/mol in case of CH2 [Citation11,Citation12]). DLPNO-CCSD(T) can be used to approximate closely the results of canonical coupled-cluster when the latter is not applicable. For instance, we have recently shown that DLPNO-CCSD(T) reproduces the canonical singlet–triplet splitting for a number of aryl-carbenes (the AC-12 set) with a root-mean-squared (RMS) error of only 0.4 kcal/mol [Citation10]. Furthermore, DLPNO calculations can be used to decompose the reference and correlation energy of bonded and non-bonded carbenes [Citation13]. This is achieved by the application of the local energy decomposition scheme (LED) for restricted and unrestricted DLPNO-CCSD [Citation14,Citation15]. However, lower-cost wave function methods or even more practically DFT calculations, despite being characterised by inconsistent behaviour [Citation10,Citation16], should in principle remain a practical tool for geometry optimizations and at least for exploratory calculations of singlet–triplet gaps, provided the performance of different functionals is carefully assessed and evaluated.
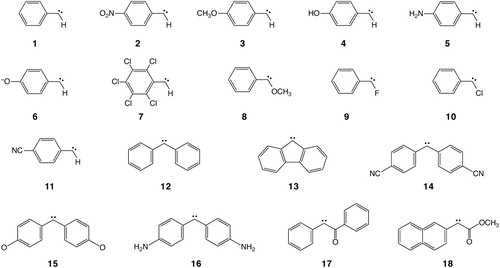
In this work we take advantage of the DLPNO approach in order to gauge the performance of a number of widely used density functionals and of orbital-optimised second-order Møller–Plesset perturbation theory (OO-MP2) and its spin-component-scaled variant (OO-SCS-MP2) [Citation17–22]. For this purpose, we have set up a benchmark to examine the performance of the tested methods both in predicting geometries and singlet–triplet gaps. 18 different aryl-carbenes were selected (AC-18 set, ) slightly extending our previous AC-12 benchmark set with larger molecules. This set is a mixture of singlet and triplet ground state aryl- and diaryl-carbenes with different electron-donating and electron-withdrawing substituents stabilising different spin states. Using the DLPNO-based coupled cluster calculations, we first analyse the quality of geometry optimisation provided by the methods evaluated in this article. Although geometry optimizations at the DLPNO-CCSD(T) using analytic gradients are not yet feasible, the total DLPNO-CCSD(T) energy at DFT and MP2 optimised geometries is an adequate measure of how closely the DFT optimisation may approach the DLPNO-CCSD(T) minimum—provided of course that the same local minimum is targeted. Hence, we take a lower total DLPNO-CCSD(T) energy as a criterion for assuming that a given DFT geometry is closer to the (unknown) DLPNO-CCSD(T) minimum than DFT derived structures that lead to higher DLPNO-CCSD(T) energies. Furthermore, critical carbene geometrical features such as carbene bond, angle and dihedral angles are analysed both chemically, in terms of substituent dependency, and computationally, in terms of method dependency. In a second step, we focus on the singlet–triplet gaps. First, a discussion about the approximations used in DLPNO coupled cluster is provided in view of the recent development of the iterative perturbative triples excitation correction that is more accurate than the previously employed semi-canonical approximation in the case of small HOMO–LUMO gaps. Finally, the accuracy of singlet–triplet gap prediction by DFT functionals is evaluated both at the structures optimised by each DFT functional itself (relaxed) and for specific reference geometries (unrelaxed) .
Figure 2. The AC-18 test set of aryl-carbenes used in this study.
2. Computational methods
Geometries were optimised using a set of DFT functionals (listed in Table ), orbital-optimised second-order Møller–Plesset perturbation theory (OO-MP2), derived by minimisation of Hylleraas functional, and its spin-component-scaled implementation OO-SCS-MP2 [Citation17–19]. Previous studies suggested that conventional UHF MP2 is unreliable for carbenes due to spin contamination of the triplet state [Citation10], therefore only the orbital-optimised variant is discussed here. Both OO-MP2 and OO-SCS-MP2 methods are employed with the resolution of the identity approximation (RI-MP2). SCS-MP2 is a variant of MP2 where the correlation energy is partitioned into same-spin and opposite-spin components that are scaled separately to achieve better performance [Citation23]. DFT calculations benefitting from the D3 dispersion correction [Citation24] with Becke–Johnson damping [Citation25] are denoted by ‘D3BJ’ [Citation25,Citation26]. All geometry optimizations have used the def2-TZVPP [Citation27] basis set and the resolution of identity approximation (RI) for Coulomb integrals together with the def2/J [Citation28] auxiliary basis sets. The chain-of-spheres approximation (COSX) to exact exchange was used in geometry optimizations with hybrid functionals [Citation29]. All integration grids were increased substantially with respect to the default values (Grid6 and GridX8 in ORCA convention). The auxiliary basis sets used set in MP2 calculations was the def2-TZVPP/C [Citation30]. Final energies for DFT methods were computed using the def2-QZVPP basis sets without the RI approximation in order to eliminate any numerical errors. SCF convergence was set to ‘VeryTight’ for all calculations reported in the present work.
Table 1. DFT functionals used in this work.
DLPNO coupled-cluster calculations for singlet and triplet states [Citation7–9] were performed either using tight or normal PNO thresholds. In the initial evaluation of geometries (Section 3.1) the relevant DLPNO calculations employed normal (default) PNO thresholds and made use of the (T0) triples corrections. Final evaluation of energies (Section 3.2) and all reference values for the singlet–triplet gaps reported in this work used tight PNO thresholds and (T1) triples. The correlation consistent basis sets cc-pVTZ and cc-pVQZ were used for the DLPNO coupled-cluster calculations [Citation31], in combination with their respective auxiliary basis sets. In DLPNO calculations the keyword ‘Randomize 0’ was used in order to ensure the consistency of localised orbitals in different calculations. We define the S-T gap as ΔEST = ET − ES, therefore negative values of splitting, when mentioned, mean a triplet ground state. All calculations were performed with ORCA [Citation32].
3. Results and discussion
3.1. Evaluation of optimised geometries
The quality of geometries optimised using OO-MP2, OO-SCS-MP2, and various DFT functionals are evaluated by calculating the DLPNO-CCSD(T0)/cc-pVTZ energies of the optimised geometries. This approach is based on the assumption that the optimum structure will have the lowest DLPNO-CCSD(T0) energy. Considering the high number of optimised geometries with various methods, one can assume that one of the optimised geometries will be sufficiently close to the optimal DLPNO-CCSD(T0)/cc-pVTZ structure to give a good qualitative overall picture. Therefore, those structures are selected as the reference structures for subsequent evaluations. Each spin state is treated separately in terms of which method performs best, and hence which method provides the reference structure. It is noted that for the initial evaluation of geometries the semi-canonical triples were used in the coupled-cluster calculations. As shown in Table , TPSS and TPSSh provide five of the reference structures, while B2GP-PLYP-D3BJ, X3LYP and B2PLYP-D3 each provide one reference structure. The rest of the lowest-energy structures are all provided by OO-SCS-MP2.
Table 2. Methods that provide the reference structure for each spin state of the carbenes considered in the present study, as evaluated using DLPNO-CCSD(T0) energies.
Table shows the absolute average energy difference between different structures to the minimum energy found among all available structures for a specific spin state of a particular species, along with the maximum deviation observed and the species for which it was observed. We note that the RMS deviation in going from NormalPNO to TightPNO thresholds is about 0.13 kcal/mol for the AC-12 benchmark subset that was reported previously [Citation10].
Table 3. Average and maximum absolute errors (kcal/mol) for each evaluated method, defined as DLPNO-CCSD(T0)/cc-pVTZ energy differences between the AC-18 carbene geometries optimized by a given method and the reference geometry that yields the lowest DLPNO-CCSD(T0) energy for each carbene in each spin state.
Inspection of Table reveals that the average errors are mostly below 1 kcal/mol. Maximum errors are, with very few exceptions, only slightly higher (ca. 2–3 kcal/mol). O3LYP produces structures with the largest deviation from the optimal minimum-energy DLPNO-CCSD(T0) structure for both singlet and triplet states. Non-hybrid functionals typically produce worse results for the singlet carbene structure compared to the triplet. However, in the case of the triplet state, the results vary significantly. Interestingly, both TPSS and TPSSh produce very good triplet geometries as judged from these energy differences, however, for the singlet geometries, TPSSh clearly surpasses the non-hybrid TPSS. Most hybrid functionals provide similar accuracies, with the exception of O3LYP as mentioned above. TPSSh provides the second-best results overall, which is important because it surpasses the performance of double-hybrid functionals at lower computational cost and better scaling. As for TPSSh, the accuracy of the geometries provided for both spin states are similar for most hybrid functionals. This can be a significant advantage because it can result in beneficial error cancelation in the calculation of S-T gaps. With regards to double-hybrid functionals, the original B2PLYP outperforms the other double hybrids included in the present work and provides very balanced results for both spin states with respect to DLPNO-CCSD(T0) energies. Despite that, double-hybrid functionals do not specifically improve upon the conventional hybrid functionals on an average sense, even though the maximum errors are reduced. Taking note of this fact and considering the higher cost of double hybrid calculations, one may conclude that application of double hybrid functionals for geometry optimizations of carbenes may not offer any advantages when it comes to creating reference geometries for the most reliable subsequent evaluation of accurate energetics.
At this point, it is useful to discuss the effect of the dispersion correction term in geometry optimisation. We focus on two functionals, B2GP-PLYP and B3LYP, with which we have optimised all structures in the AC-18 set with and without the D3BJ dispersion correction. The difference in both cases is insignificant and the small changes effected by the empirical dispersion correction do not always improve the structures. Although one may not necessarily expect a considerable advantage from including dispersion effects for the carbene molecules of this study, the above results suggest that empirical corrections such as the D3BJ approach employed here are not beneficial for medium-sized carbene systems on an average sense.
Having discussed the performance of DFT functionals, we now turn to the orbital-optimised OO-MP2 and OO-SCS-MP2 methods. The orbital-optimised variant of the MP2 method is selected as it is remedying the spin contamination of the reference wavefunction [Citation10]. Spin component scaling empirically counters the bias toward same spin electrons of the SCF calculations by adjusting the contribution of same-spin and opposite-spin electron correlation. The overestimation of non-bonding interactions by MP2 is also suggested to be remedied by the SCS modification [Citation24]. For the present comparison against DLPNO-CCSD(T0) energies, OO-MP2 provides relatively good geometries especially for the triplet states, however OO-SCS-MP2 improves on the OO-MP2 structures in all cases but one, thus providing the optimal reference structures. Therefore, between the two options we can recommend uniformly the application of the OO-SCS-MP2 variant. It has been noted that OO-MP2 may tend to overestimate correlation effects with concomitant effects on bond lengths [Citation17,Citation53], an effect that could be damped in the SCS variant consistent with the results obtained here. Although not tested in the present work, it is also worth mentioning the regularised OO-MP2 approach of Head-Gordon and co-workers [Citation54,Citation55], which can successfully counter divergences that may occur during the optimisation procedure for OO-MP2 when the frontier orbitals approach degeneracy.
Despite the above findings, considering the relatively high cost of orbital-optimised MP2 compared to any DFT alternative, it is recognised that depending on the type of application it may be questionable whether the higher computational cost always justifies the increase in accuracy, particularly given the good performance of standard hybrid functionals. Therefore, as an alternative to OO-SCS-MP2, the recommended DFT approach for obtaining reliable structures is TPSSh. Figure provides a closer look at the quality of the structures provided by TPSSh per species of the AC-18 set. The most problematic cases in terms of unfavourable DLPNO-CCSD(T0) energies seem to be species 14, 17, and 18. Interestingly, these are all species which feature a distinct deviation from planarity between the two carbene substituents. Therefore, this issue is likely related to the correlated prediction of correct carbene bond distance, carbene angle, and dihedral angle between the carbene substituents. In the following, we take a closer look individually at these structural parameters.
Figure 3. Energy errors (kcal/mol) of TPSSh-optimized geometries for the carbenes of the AC-18 set. The energy error is defined as the DLPNO-CCSD(T0) energy difference between the TPSSh geometry and the optimized geometry that provides the lowest DLPNO-CCSD(T0) energy for each carbene in each spin state.
3.1.1. Carbene bond length
The variation in the length of the C–C bond between the carbene centre and the phenyl group in substituted carbenes may be qualitatively connected to Bent’s rule [Citation56], according to which the σ bond length decreases as s orbital character increases. This would imply that the C–C bonds involving the carbene centre might be shorter in the singlet state because this features a smaller carbene angle and, hence, larger s character in the carbene sp2 orbital. However, the opposite is observed, with the triplet state having shorter C–C bonds. The longer bond length in the singlet state is presumably due to the additional electron repulsion between the σ-bond substituents and the doubly occupied σ-sp2 orbital [Citation57]. It is also noted that chemical substitution of stabilising groups for each spin state reduces the bond length (electron donors for singlet and electron withdrawal groups for the triplet carbenes).
Deviations in the optimised bond lengths of each method in comparison to the overall optimal reference structure are shown in Figure . All DFT functionals and wave function-based methods predict bond lengths within 0.03 Å of the optimal, the only exception being the TPSS-optimised carbene–carbon bond in species 2 and 11. This seems to be an anomaly as the TPSS singlet wavefunction is spin-contaminated (specifically, <S2> = 0.84 and 0.79 for carbenes 2 and 11, respectively) because it converges to a broken-symmetry state (‘open shell’ singlet). This issue occurs also with TPSSh but to a much smaller extent. No other functionals showed such behaviour for these two or any other carbenes.
Figure 4. Average and maximum errors of tested methods in carbene–carbon bond lengths (in Å) against the minimum DLPNO-CCSD(T0) energy reference structures.
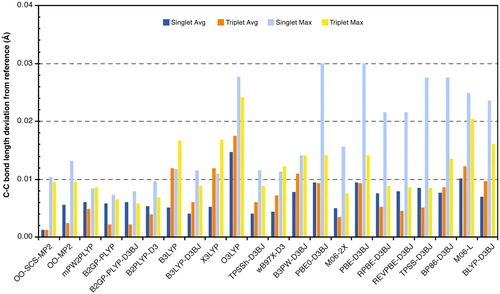
In general, the deviations among methods are not pronounced. Most methods slightly underestimate the C–C bond length compared to the reference geometry. In the singlet state, the results present a wider scatter between different GGA, hybrid, and double hybrid methods compared to the triplet state. Most hybrid and GGA functionals provide bond lengths within 0.01 Å for the singlet state, with the exception of O3LYP that yields the shortest bond lengths among all methods for both spin states. In the triplet state, most of the hybrid and GGA functionals show larger deviations from the reference geometry. However, TPSSh provides on average results that are balanced for both spin states and close in comparison to the more expensive wave function-based and double hybrid methods. The Minnesota hybrid M06-2X is the opposite extreme compared to O3LYP, as it provides the longest bond lengths for the singlet, in this respect better-resembling OO-SCS-MP2. Double hybrid functionals and M06-2X also yield in general longer bond lengths than the GGA and hybrid functionals in the triplet state, which is closer to the MP2-based methods.
3.1.2. Carbene angle
The carbene angle is the most critical structural parameter of carbenes. If a carbene was linear then both π and σ orbitals would be degenerate, resulting in a triplet ground state. As the carbene centre bends, the σ orbital gains s character and relaxes. A competition between electron–electron repulsion and orbital relaxation then determines the carbene angle. Therefore, singlet states have a more bent structure to maximise orbital relaxation and reduce degeneracy. Prediction of carbene angle presents a challenging problem for most methods in specific aryl-carbenes. In the singlet state, most species bonded with a single phenyl ring provide acceptable angles with a maximum error below 5 degrees. Addition of a second phenyl ring slightly decreases the accuracy. Considerable errors are observed for species 17, where errors up to 10 degrees are observed for the typically most accurate method OO-SCS-MP2. Table lists deviations from the structure providing the lowest energy at coupled-cluster level for species 17 (in this case, B2GP-PLYP-D3BJ). As can be seen, orbital-optimised MP2 methods show significant divergence from the optimum angle as judged by minimal DLPNO-CCSD(T0) energies. Double hybrid methods on the other hand provide uniformly very good angles.
Table 4. Absolute errors in optimized carbene angles (in degrees) for species 17 in its singlet state and species 14 in its triplet state.
Calculation of the optimal angle of the triplet state appears to be even more challenging for most methods. Overall maximum errors are higher in the triplet state in comparison to the singlet state. The most problematic species in the triplet state is 14, where the maximum error can be up to 8 degrees as shown in Table . In this case, the only straightforward solution in achieving the optimum angle is the use of a double hybrid functional or orbital-optimised MP2. The results of these methods are approximated very well by M06-2X. It is noted that the addition of exact exchange does not necessarily improve the performance of GGA functionals, as judged by comparison of related pairs of hybrid and non-hybrid functionals.
Figure shows the absolute average error of all methods used in this study. OO-SCS-MP2 provides the best overall carbene angles although it faces large errors in the specific case of the singlet state of species 17. As was evident from the detailed analysis of the worst-case scenarios of both spin states, the addition of exact exchange does not clearly improve the prediction of carbene angle. However, all methods benefiting from perturbation theory provide highly accurate carbene angles. Despite the occasional large errors in specific cases, absolute average errors can be considered small.
Figure 5. Absolute average and maximum errors in carbene angles (in degrees) of tested methods against the minimum DLPNO-CCSD(T0) energy reference structures.
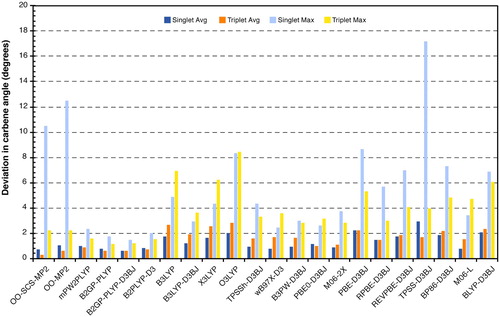
We note that the carbene angle is not dependent on the empty p orbital, therefore, it is not considerably affected by the substitution of electron donor or acceptor groups. This is not the case for the out-of-plane distortions that will be discussed in the following.
3.1.3. Carbene dihedral angle
As mentioned above, dihedral angles involving the carbene site seem to be among the trickiest geometrical parameters to predict correctly. The out-of-plane conformation of the groups attached to the carbene centre is affected by steric and electronic effects. Principally the dihedral angle is determined by the steric demands of aryl groups; addition of electron donors and acceptors to the aromatic ring introduces further modifications. The phenyl rings in diaryl-carbenes are always forced out of a plane in both spin states. A few substituted aryl-carbene species also contribute in tilting the substituent at the carbene centre out of the plane of the other phenyl ring, such as with species 18. These twists are spin-state specific and may be suppressed or entirely absent in one of the two spin states. Specifically, for substituent effects, the dihedral angle appears to correlate with change in the effective electron donation or withdrawal to the phenyl ring (resonance adjustment) and when this happens the dihedral angle can be significantly different between the two spin states. The more in-plane conformations increase the magnitude of the effect of the ring substituent and the opposite occurs for more out-of-plane conformations [Citation58].
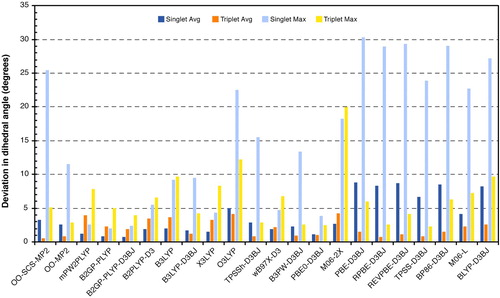
As seen from the results in Figure , the absolute average error for all methods in the value of the dihedral angle is below 6 degrees. Local functionals are characterised by large deviations in the singlet state. This seems to be the primary reason for the considerably poor performance of GGA functionals for the singlet state. Surprisingly, double hybrid methods produce worse triplet dihedral angles than hybrid and GGA functionals. This might be related to MP2 problems with spin contamination. Orbital-optimised MP2 methods have rather large average errors in the singlet state, however this is mostly due to a large error in a single species (17), where the dihedral angle is about 25 degrees off. Otherwise, the orbital-optimised MP2-based methods provide accurate dihedral angles for all the other carbenes of the test set.
Figure 6. Absolute average and maximum errors in carbene dihedral angles (in degrees) of tested methods against the minimum DLPNO-CCSD(T0) energy reference structures.
3.1.4. Qualitative analysis of dihedral change by substituent
It is informative to take a closer look at specific cases for a qualitative discussion of variation of dihedral angle by the presence of electron donor and acceptor groups on the aryl rings. Focusing first on species 2, the triplet state is stabilised by the electron-withdrawing nitro group. The nitro group helps to restore the resonance of the phenyl ring that is reduced in the triplet state by delocalisation of the carbene p electron on the aryl. However, this effect is preferentially reduced in the singlet state so the phenyl ring can instead donate electron density to the empty p orbital on the carbene centre. Therefore, in the singlet state, the nitro group is slightly twisted out of plane to reduce the electron-withdrawing effect. Prediction of the twisted singlet state varies significantly among different methods and a twist of up to 20–30 degrees is predicted by most GGA methods. However, the reference structure (B2PLYP-D3) predicts a more modest out-of-plane angle of ca. 5 degrees. In the case of the para-substitution with an amine group (species 5) both amine hydrogens are bent out of the aryl ring plane in the same direction for both spin states. The bending is more pronounced in the singlet state. This can be rationalised by the different extent of conjugation of the nonbonding electron pair of the nitrogen with the aromatic system of the phenyl, depending on the occupation of the carbene p orbital and its coupling through the π-network of the benzene ring.
The methoxy group bound at the carbene centre in species 8 forms a planar structure in the singlet state but the methyl bends out of plane in the triplet state. This can be attributed to the interaction between the oxygen lone pairs with the unpaired electrons in the triplet carbene, whereas the singlet state is in-plane to maximise the π–π interaction between the oxygen lone pair and the empty p orbital of carbene. Prediction of this twist is overall quite accurate, with errors within a few degrees for most methods.
Substitution of electron donor and acceptor into the diphenylcarbene results to small effects, with a single exception of O-. All cases follow the same trend mentioned before, the electron-donating group O- significantly increases the dihedral angle in the triplet state, while in the singlet state the dihedral angle is significantly reduced to increase electron donation. Electron withdrawing groups CN and NH2 have a much smaller structural effect. In the case of species 17 and 18, where the carbene centre is adjacent to a carbon atom bonded to one or two oxygen atoms, the triplet and singlet states have significantly different structures. In the singlet state, the structure is bent so the empty p orbital can interact with the oxygen electron pair. However, the triplet state adopts a planar structure.
3.2. Singlet–triplet gaps
3.2.1. Approximations relevant to the reference calculations
In DLPNO coupled-cluster calculations, the reference energy value (restricted HF for the singlet, quasi-restricted for the triplet) is the same as in the canonical counterpart. However, there is a deviation in the value of the correlation energy because the virtual space is localised in order to enable the lower scaling that characterises the DLPNO approach. We call this deviation ‘localisation error’. Table lists deviations for the different excitation components of coupled-cluster theory and additionally compares deviations due to distinct approaches to the triples corrections. As can be seen, the error introduced by the DLPNO approximation is very small at the CCSD level for most cases, with the maximum deviation observed for species 17, the only case exceeding 0.5 kcal/mol. In the case of triple excitations, since DLPNO calculations use quasi-restricted orbitals, in canonical coupled-cluster theory one needs to either iteratively calculate the perturbative triples amplitudes or diagonalise the internal and virtual spaces separately. The latter case is not possible in case of localised orbitals. This means one either ignores the off-diagonal elements, i.e. the (T0) approximation, or iteratively calculates the perturbative triple excitation amplitudes, i.e. (T1) [Citation59]. From Table , it is clear that the (T0) approximation may have a non-negligible effect on singlet–triplet gaps, however (T1) only slightly suffers from the localization error, to an extent usually less than the associated CCSD error. The reference values for this work were all obtained with perturbative (T1) triples.
Table 5. Deviations in singlet–triplet gaps (kcal/mol) for various comparisons of approaches relevant to the DLPNO-CCSD(T) calculations.
3.2.2. Singlet–Triplet gaps by DFT methods
To compare the accuracy of DFT methods for spin-state energetics, we have considered two different approaches that represent likely scenarios for practical applications: (a) singlet–triplet gaps based on structures optimised with each specific method, and (b) singlet–triplet gaps computed with single-point calculations using a common set of reference structures. In the present case, these common structures were obtained with OO-SCS-MP2. The reference singlet–triplet gaps in both cases are the same and are obtained using DLPNO-CCSD(T1) energies with cc-pVTZ/cc-pVQZ basis set extrapolation on the OO-SCS-MP2 optimised geometries. In the following we will focus on the overall average errors of the methods without going into details for individual carbenes of the test set. The results are presented in Table . Detailed results are provided as Supporting Material.
Table 6. Evaluation of density functional theory methods for singlet–triplet gaps using either geometries optimized individually with each functional, or reference OO-SCS-MP2 geometries: absolute average errors (AAE), root-mean-squared errors (RMS), and maximum errors (Max) in kcal/mol against reference cc-pV(T/Q)Z basis-set-extrapolated DLPNO-CCSD(T1) calculations.
A crucial observation we would like to highlight from the outset is that the functionals which provide good structures as discussed in the preceding sections of this paper, do not necessarily provide good energetics according to the results of Table . Most prominently, TPSSh provides some of the worst singlet–triplet gaps whilst providing very good geometries across the AC-18 set. Therefore, it matters a great deal for practical applications whether the target is a high-quality geometry that can be the basis for further investigations with higher-level methods, or whether the target is a reliable screening for singlet–triplet gaps.
Most GGA functionals provide singlet–triplet gaps with average errors of more than 2 kcal/mol and maximum errors of over 6 kcal/mol. Interestingly BLYP, one of the cheapest methods assessed here, provides very reasonable average errors of less than 2 kcal/mol, with a maximum error of less than 6 kcal/mol. The functional is directly competitive to hybrid functionals such as B3LYP and X3LYP, which do not uniformly improve on BLYP over the whole set. An interesting observation is that the D3BJ dispersion corrected version of B3LYP, which is the best hybrid functional among the teste methods, performs better for singlet–triplet gaps than the uncorrected form even when using the OO-SCS-MP2 geometries. Double-hybrid methods, specifically mPW2PLYP and B2PLYP, can be recommended as the best possible choices among DFT methods. It is important to note that on average they do not improve dramatically on the singlet–triplet gaps obtained by, for example, the simple BLYP functional. However, they achieve much better error distribution. The results change little when considering singlet–triplet gaps calculated using OO-SCS-MP2 structures. In fact, the average and maximum errors are generally increased, but a gain is observed when using OO-SCS-MP2 geometries for mPW2PLYP and TPSS.
Overall, the above results suggest that BLYP is a fine choice at least for exploratory calculations on singlet–triplet gaps, whereas a conceivable composite DFT-based method that should result in both, better geometries and better energies, might combine TPSSh for geometry optimisation and mPW2PLYP for calculation of final energies.
A final comment is in order with regard to the expected accuracy of DFT methods versus wave function based approximations. OO-SCS-MP2 and OO-MP2 provide RMS errors of 3.82 and 3.49 kcal/mol for singlet–triplet gaps (AAE of 3.27 and 2.29 kcal/mol, respectively) using self-consistently optimised geometries. The average error of OO-MP2 is practically the same when it used with OO-SCS-MP2 geometries (AAE and RMS of 2.25 and 3.33 kcal/mol, respectively). The typical errors of the two methods are within 2–3 kcal/mol of the reference in most cases. A prominent exception is the challenging carbene 17, for which they both exhibit maximum absolute errors of over 10 kcal/mol compared to the reference, predicting an inverse S-T gap, i.e. a singlet ground state. A consistent difference among the two is that OO-SCS-MP2 favours the singlet state in comparison to OO-MP2, but it is likely that there is overcompensation of the triplet preference of OO-MP2, resulting in slightly worse singlet–triplet gaps on average for the SCS variant. An interesting comparison can be made with the restricted open-shell version of MP2 (RO-MP2). The performance of RO-MP2 and OO-MP2 is very similar (see Supporting Material for explicit comparisons of S-T gaps at OO-SCS-MP2 geometries), but the former improves on average over the latter with AAE and RMS errors of 1.48 and 1.60 kcal/mol, respectively. Importantly, RO-MP2 correctly predicts a triplet ground state for carbene 17, with an S-T gap within 1.5 kcal/mol of the reference. This is not the only source of improvement, however, because RO-MP2 has a slight edge over OO-MP2 even when this specific carbene is excluded from the comparison (AAE of 1.48 vs 1.69 kcal/mol). Overall, it can be concluded that MP2 methods do not match the performance of double hybrid functionals, and are surprisingly challenged even by a functional such as BLYP.
Turning to coupled-cluster approaches, it is noted that the AAE, RMS and Max values for CCSD, i.e. with the exclusion of any triples corrections, are 1.43, 1.57, and 3.90 kcal/mol using the OO-SCS-MP2 geometries. Considering the results presented in Table , these average metrics are comparable with, but inferior to the results of double-hybrid methods, therefore the choice of CCSD over mPW2PLYP would be questionable. A systematic improvement on singlet–triplet gaps over double-hybrid DFT can therefore only be achieved with inclusion of perturbative triples to CCSD.
4. Conclusion
A range of DFT methods and orbital-optimised versions of MP2 and SCS-MP2 were evaluated for geometric parameters and singlet–triplet gaps of aryl-carbenes against DLPNO-based coupled cluster calculations. In the case of geometry optimisation, we found that TPSSh is the most practical method that provides accurate structures at a reasonable cost. If the system size allows the application of more expensive methods, OO-SCS-MP2 provides the most accurate structures as judged against DLPNO-CCSD(T) energetics. In case of very large systems, the faster non-hybrid TPSS could be the method of choice for good geometries. Further investigation into geometrical parameters of carbenes demonstrates that the in- and out-of-plane dihedral rotations are the most challenging parameters to capture in the aryl-carbene series. Crucially, these parameters have a significant energetic consequence due to resonance stabilisation of the carbene centre. The accuracy of DFT functionals in calculation of singlet–triplet gaps is mixed. Importantly, using better or worse structures does not significantly change on average the quality of singlet–triplet gaps computed by DFT functionals. Interestingly, BLYP provides very good singlet–triplet energy gaps and we recommend this functional for exploratory calculations and the mPW2PLYP double hybrid or DLPNO-CCSD(T) when higher accuracy is desired.
TMPH-2020-0107-File004.pdf
Download PDF (180.3 KB)Acknowledgement
Support by the Max Planck Society is gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- W. Kirmse, Carbene Chemistry, 2nd ed. (Academic Press, New York, 1971).
- G. Bertrand, Carbene Chemistry: From Fleeting Intermediates to Powerful Reagents. (CRC Press, Boca Raton, 2002), p. 320.
- Y. Wang, C.M. Hadad and J.P. Toscano, J. Am. Chem. Soc. 124, 1761–1767 (2002). doi: 10.1021/ja012139v
- P. Costa and W. Sander, Angew. Chem., Int. Ed. 53, 5122–5125 (2014).
- S. Henkel, P. Costa, L. Klute, P. Sokkar, M. Fernandez-Oliva, W. Thiel, E. Sanchez-Garcia and W. Sander, J. Am. Chem. Soc. 138, 1689–1697 (2016). doi: 10.1021/jacs.5b12726
- Z. Zhu, T. Bally, L.L. Stracener and R.J. McMahon, J. Am. Chem. Soc. 121, 2863–2874 (1999). doi: 10.1021/ja983277w
- C. Riplinger and F. Neese, J. Chem. Phys. 138, 034106 (2013). doi: 10.1063/1.4773581
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys. 139, 134101 (2013). doi: 10.1063/1.4821834
- M. Saitow, U. Becker, C. Riplinger, E.F. Valeev and F. Neese, J. Chem. Phys. 146, 164105 (2017). doi: 10.1063/1.4981521
- R. Ghafarian Shirazi, F. Neese and D.A. Pantazis, J. Chem. Theory Comput. 14, 4733–4746 (2018). doi: 10.1021/acs.jctc.8b00587
- J.F. Harrison, in Advances in the Theory of Atomic and Molecular Systems, edited by P. Piecuch, J. Maruani, G. Delgado-Barrio and S. Wilson (Springer, Dordrecht, 2009), pp. 33–43.
- H.F. Schaefer, Science. 231, 1100–1107 (1986). doi: 10.1126/science.231.4742.1100
- R. Ghafarian Shirazi, F. Neese, D.A. Pantazis and G. Bistoni, J. Phys. Chem. A. 123, 5081–5090 (2019). doi: 10.1021/acs.jpca.9b01051
- W.B. Schneider, G. Bistoni, M. Sparta, M. Saitow, C. Riplinger, A.A. Auer and F. Neese, J. Chem. Theory Comput. 12, 4778–4792 (2016). doi: 10.1021/acs.jctc.6b00523
- A. Altun, M. Saitow, F. Neese and G. Bistoni, J. Chem. Theory Comput. 15, 1616–1632 (2019). doi: 10.1021/acs.jctc.8b01145
- H.L. Woodcock, D. Moran, B.R. Brooks, P.v.R. Schleyer and H.F. Schaefer, J. Am. Chem. Soc. 129, 3763–3770 (2007). doi: 10.1021/ja068899t
- R.C. Lochan and M. Head-Gordon, J. Chem. Phys. 126, 164101 (2007). doi: 10.1063/1.2718952
- F. Neese, T. Schwabe, S. Kossmann, B. Schirmer and S. Grimme, J. Chem. Theory Comput. 5, 3060–3073 (2009). doi: 10.1021/ct9003299
- S. Kossmann and F. Neese, J. Phys. Chem. A. 114, 11768–11781 (2010). doi: 10.1021/jp105647c
- U. Bozkaya, J.M. Turney, Y. Yamaguchi, H.F. Schaefer, 3rd and C.D. Sherrill, J. Chem. Phys. 135, 104103 (2011). doi: 10.1063/1.3631129
- U. Bozkaya, J. Chem. Theory Comput. 10, 4389–4399 (2014). doi: 10.1021/ct500634s
- U. Bozkaya, J. Chem. Theory Comput. 10, 2371–2378 (2014). doi: 10.1021/ct500231c
- T.P.M. Goumans, A.W. Ehlers, K. Lammertsma, E.-U. Würthwein and S. Grimme, Chem. Eur. J. 10, 6468–6475 (2004). doi: 10.1002/chem.200400250
- O. Marchetti and H.-J. Werner, J. Phys. Chem. A. 113, 11580–11585 (2009). doi: 10.1021/jp9059467
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem. 32, 1456–1465 (2011). doi: 10.1002/jcc.21759
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys. 132, 154104 (2010). doi: 10.1063/1.3382344
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys. 7, 3297–3305 (2005). doi: 10.1039/b508541a
- F. Weigend, Phys. Chem. Chem. Phys. 8, 1057–1065 (2006). doi: 10.1039/b515623h
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys. 356, 98–109 (2009). doi: 10.1016/j.chemphys.2008.10.036
- A. Hellweg, C. Hättig, S. Höfener and W. Klopper, Theor. Chem. Acc. 117, 587–597 (2007). doi: 10.1007/s00214-007-0250-5
- T.H. Dunning, Jr, J. Chem. Phys. 90, 1007–1023 (1989). doi: 10.1063/1.456153
- F. Neese, WIRES Comput. Mol. Sci. 8, e1327 (2018). doi: 10.1002/wcms.1327
- A.D. Becke, Phys. Rev. A. 38, 3098–3100 (1988). doi: 10.1103/PhysRevA.38.3098
- J.P. Perdew, Phys. Rev. B. 33, 8822–8824 (1986). doi: 10.1103/PhysRevB.33.8822
- C. Lee, W. Yang and R.G. Parr., Phys. Rev. B. 37, 785–789 (1988). doi: 10.1103/PhysRevB.37.785
- J.P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett. 77, 3865–3868 (1996). doi: 10.1103/PhysRevLett.77.3865
- Y. Zhang and W. Yang, Phys. Rev. Lett. 80, 890–890 (1998). doi: 10.1103/PhysRevLett.80.890
- J.P. Perdew and Y. Wang, Phys. Rev. B. 45, 13244–13249 (1992). doi: 10.1103/PhysRevB.45.13244
- B. Hammer, L.B. Hansen and J.K. Nørskov, Phys. Rev. B. 59, 7413–7421 (1999). doi: 10.1103/PhysRevB.59.7413
- A.D. Becke, J. Chem. Phys. 98, 1372–1377 (1993). doi: 10.1063/1.464304
- W.-M. Hoe, A.J. Cohen and N.C. Handy, Chem. Phys. Lett. 341, 319–328 (2001). doi: 10.1016/S0009-2614(01)00581-4
- X. Xu and W.A. Goddard, Proc. Natl. Acad. Sci. U. S. A. 101, 2673–2677 (2004). doi: 10.1073/pnas.0308730100
- C. Adamo and V. Barone, J. Chem. Phys. 110, 6158–6170 (1999). doi: 10.1063/1.478522
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys. 128, 084106 (2008). doi: 10.1063/1.2834918
- J. Tao, J.P. Perdew, V.N. Staroverov and G.E. Scuseria, Phys. Rev. Lett. 91, 146401 (2003). doi: 10.1103/PhysRevLett.91.146401
- Y. Zhao and D.G. Truhlar, J. Chem. Phys. 125, 194101 (2006). doi: 10.1063/1.2370993
- V.N. Staroverov, G.E. Scuseria, J. Tao and J.P. Perdew, J. Chem. Phys. 119, 12129–12137 (2003). doi: 10.1063/1.1626543
- Y. Zhao and D.G. Truhlar, Theor. Chem. Acc. 120, 215–241 (2008). doi: 10.1007/s00214-007-0310-x
- S. Grimme, J. Chem. Phys. 124, 034108 (2006). doi: 10.1063/1.2148954
- A. Karton, A. Tarnopolsky, J.-F. Lamère, G.C. Schatz and J.M.L. Martin, J. Phys. Chem. A. 112, 12868–12886 (2008). doi: 10.1021/jp801805p
- C. Adamo and V. Barone, J. Chem. Phys. 108, 664–675 (1998). doi: 10.1063/1.475428
- T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys. 8, 4398–4401 (2006). doi: 10.1039/b608478h
- M. Marsman, A. Grüneis, J. Paier and G. Kresse, J. Chem. Phys. 130, 184103 (2009). doi: 10.1063/1.3126249
- D. Stück and M. Head-Gordon, J. Chem. Phys. 139, 244109 (2013). doi: 10.1063/1.4851816
- J. Lee and M. Head-Gordon, J. Chem. Theory Comput. 14, 5203–5219 (2018). doi: 10.1021/acs.jctc.8b00731
- H.A. Bent, Chem. Rev. 61, 275–311 (1961). doi: 10.1021/cr60211a005
- Y. Xie, P.R. Schreiner, P.v.R. Schleyer and H.F. Schaefer, J. Am. Chem. Soc. 119, 1370–1377 (1997). doi: 10.1021/ja9517072
- M.A. Dobrowolski, T.M. Krygowski and M.K. Cyrański, Croat. Chem. Acta. 82, 139–147 (2009).
- Y. Guo, C. Riplinger, U. Becker, D.G. Liakos, Y. Minenkov, L. Cavallo and F. Neese, J. Chem. Phys. 148, 011101 (2018). doi: 10.1063/1.5011798