

Abstract
Low-lying states in small, multiply bonded hydrocarbon molecules may be divided into valence and Rydberg excitations. Excited states of the cubic phase of the acetylene molecular crystal are calculated using the time-dependent Hartree–Fock (TDHF) method. TDHF and Bethe–Salpeter equation (BSE) calculations are performed on molecular CH
in order to interpret these excited states. Computationally inexpensive TDHF and BSE methods are compared to previous CASPT2, MRCI and EOM-CCSD calculations and experiment. Localised, lowest energy excited states in the C
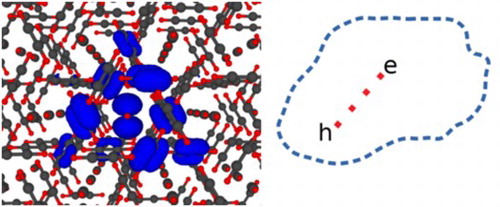
H
molecular crystal mirror those in the gas phase. In the next lowest excitations the electron and hole separate over one or two shells of molecular neighbours. A density fitting method for calculating the Coulomb matrix elements which arise in time-dependent Hartree Fock theory (TDHF) in periodic systems is implemented in the Exciton code and described briefly.
GRAPHICAL ABSTRACT
Acknowledgements
This work was supported by TCHPC (Research IT, Trinity College Dublin). Calculations were performed on the Boyle and Kelvin clusters maintained by the Trinity Centre for High Performance Computing. The Boyle cluster was funded through grants from the European Research Council and Science Foundation Ireland. The Kelvin cluster was funded through grants from the Irish Higher Education Authority, through its PRTLI programme.
Disclosure statement
No potential conflict of interest was reported by the author.
Notes
1 The TZVPPD C basis was modified by replacing the two most diffuse s exponents by one exponent of 0.129, increasing the most diffuse p exponent from 0.100 to 0.129 and removing the most diffuse d and f exponents. The TZVPPD H basis was modified by removing the most diffuse p exponent.