
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Many classes of organic reactions exhibit a remarkable increase in reaction rates when they occur at the water–organic interface. Although this observed ‘on-water’ catalysis has been extensively studied, the suggested mechanisms still do not explain some of the experimental findings. The mechanism proposed by Jung and Marcus (JACS 129, 5492 (2007)) involves stabilising the transition-state (TS) complex via H-bonds to ‘dangling’ interfacial water molecules. Although the reactants also experience H-bonding to interfacial water molecules in the reactant configuration, it has been argued that the H-bonds are enhanced, in terms of number and strength, in the TS. Therefore, the observed decrease in activation energy has been attributed to this preferential enhancement of H-bonds which leads to a more pronounced TS stabilisation. We employ energy decomposition analysis using the method of absolutely localised molecular orbitals to study this proposition. We find that H-bonds to interfacial water molecules are equivalent in the TS and reactant configurations. Nevertheless, these H-bonds result in significantly enhanced charge-transfer between the reactants in the TS complex, which rationalises the decrease in activation energy.
GRAPHICAL ABSTRACT
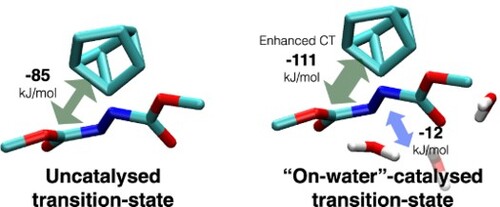
1. Introduction
The field of on-water catalysis added a greener and economic dimension to the current toolbox of organic synthesis. The term ‘on-water’ refers to reactions in which the transition-state (TS) occurs at the organic–water interface on the organic side [Citation1]. This is to differentiate in-water catalysis, which had been previously investigated and refers to reactions occurring in the bulk of the solution [Citation2]. Over the past two decades, many experimental and theoretical studies contributed to the current understanding of the on-water phenomenon [Citation2]. There are three main observations associated with on-water catalysis [Citation3]. Firstly, heterogeneity of the reaction mixture is essential. Secondly, the interface must be with water. Thirdly, some reactions tend to show a large isotope effect, while others show a very mild one. In-water catalysis has been mainly attributed to hydrophobic effects and the cohesive energy density of water [Citation2,Citation4]. On the other hand, on-water catalysis has been rationalised using two mechanisms. The first mechanism involves lowering of the activation energy via H-bonds from dangling OH groups at the water interface [Citation4]. The presence of these dangling OH groups with extra liability to H-bonding has been supported by various experimental and theoretical studies [Citation2,Citation4]. The second catalytic mechanism, proposed by Beattie and coworkers, involves proton-transfer to the organic reactants [Citation3].
The cycloaddition reaction between quadricyclane (1) and dialkyl azodicarboxylates is a good example to investigate the mechanism of on-water catalysis (Figure ). The cycloadditions involving 1 with dimethyl azodicarboxylate (DMAzD, 2a) or diethyl azodicarboxylate (DEAzD, 2b) show different catalytic rates and isotope effects [Citation2]. Using a TS model for the reaction of 1 and 2a that forms H-bonds with the dangling OH groups at the water interface, Jung and Marcus calculated a drop in activation energy of 7.5 kcal/mol. This value is very close to the experimental value (5.4 kcal/mol) for the on-water cycloaddition of 1 and 2b [Citation5]. Nevertheless, catalysis via H-bonding does not explain the strong isotope effect of the reaction of 1 and 2a [Citation3]. Domingo et al. computed a similar fall in activation energy (8.5 kcal/mol) for the cycloaddition of 1 and 2a albeit adopting a polar mechanism [Citation6]. The mechanism by Domingo et al. is more compatible with the proton-transfer explanation of Beattie and coworkers [Citation2]. The proton-transfer mechanism explains the isotope effect observed in many on-water catalysed reactions and is consistent with the fact that most of the reactions that are catalysed on-water are also subject to acid-base catalysis [Citation3]. Being a hot topic of research, there are a plethora of experimental and theoretical studies endorsing either H-bond or proton-transfer explanations of on-water catalysis [Citation2].
Figure 1. Cycloaddition reaction of quadricyclane (1) and dialkyl azodicarboxylates (2) and three derivatives (2a, 2b and 2c).
In particular, Jung and Marcus concluded that the on-water reaction between 1 and 2a was catalysed due to extra stabilisation of the TS. In their reaction model, 2a makes three H-bonds with water in the TS configuration, while it makes only two H-bonds in the reactant one. They also computed partial charges on the nitrogens and carbonyl oxygens of 2a in the TS and reactant configurations. They deduced that 2a also forms stronger H-bonds to interfacial water molecules in the TS configuration which had more electronegative atoms than the reactant counterpart. Accordingly, they predicted that replacing the nitrogens in DMAzD (2a) with an olefin (2c) or an acetylene group with lower capability of H-bonding would result in inefficient catalysis [Citation4]. Worth noting, Beattie et al. showed experimentally that the cycloaddition of cyclopentadiene and 2c actually exhibits on-water catalysis [Citation3]. Later, Karhan et al. studied the same cycloaddition of 1 and 2a using second-generation Car–Parrinello molecular dynamics [Citation7]. Only a marginal increase of H-bonds has been noticed at the TS. The focus of this work is thus to investigate the relative strengths of H-bonds between 2a and interfacial water molecules in the reactant and TS models.
2. Computational methods
We performed energy decomposition analysis (EDA) of the reactants and TS models for the reaction of 1 and 2a. The method of absolutely localised molecular orbitals (ALMO), available in the CP2K suite (version 7.0)[Citation8], was used to decompose the interaction energy into frozen density, polarisation and charge-transfer (CT) components. Recent reviews are already available on EDA [Citation9,Citation10]. We briefly outline the ALMO EDA method, while the interested reader can find more details and applications in the cited literature [Citation11–14].
The ALMOs on a given fragment, x, are used to compute the state which is infinitely separated from all other fragments. The occupied ALMOs of each fragment are then used to yield an antisymmetrised wave functions
of the complex composed of all fragments. At that stage, the ALMOs on a given fragment are not relaxed, except by the changes needed to satisfy the Pauli exclusion principle. The frozen energy contribution is defined as the difference between the SCF energies of those two states:
(1)
(1) which means that it includes the Coulomb term as well as an exchange-correlation term. In the next step, the ALMOs on a given fragment are allowed to relax in the field of all other fragments to yield the state
The ALMOs of
are still localised at their fragments. The polarisation energy is then defined as:
(2)
(2) The final portion of interaction energy is defined as the difference between the energy of the fully delocalised state
and the polarised state:
(3)
(3) where
is the energy lowering due to the total electron transfer from the occupied ALMOs on one fragment to the virtual orbitals on the others (forward donation), in addition to the back donation due to repolarisation.
Our ALMO calculations were conducted at the M06-2x/aug-cc-pVTZ level of theory using the Gaussian-augmented plane wave all-electron approach [Citation8,Citation15–17]. Five setups where initially used in total, as shown in Figure : 2a with 3 water molecules (the 3 water molecules are collectively referred to as ‘ 3’) in the reactants configuration (i), and the TS comprised of the coordinates of 1, 2a and 3(ii). The coordinates for i and ii were taken from the study by Jung and Marcus [Citation4] and are provided in Table S1 along with key geometric elements in Figure S1 in the electronic supplementary information (ESI). The three remaining setups were derived from ii to get insight on the EDA of the interaction energy between a pair of 1, 2a and 3 in the absence of the third. This way, the role of including water molecules in the transition state complex can be discerned. The coordinates of 1 were deleted to investigate the interaction between 2a and 3 in the TS conformation (iii). Similarly, the coordinates of 3 were deleted from ii to study the interaction between 1 and 2a maintaining the TS conformation (iv). Finally, the coordinates of 2a were deleted from (ii) to investigate possible interactions between 1 and 3 in the TS conformation (v). Alternative partitioning of the system, taking the three water molecules as a single fragment and thus excluding the inter-water energy components, is provided in Figure .
Figure 2. The five systems studied by EDA: (i) reactant conformation of 2a and 3, (ii) TS, (iii) TS without 1, (iv) TS without 3 and (v) TS without 2a. The CT between a given pair of fragments is indicated by an arrow and the corresponding value is given in kJ/mol. Water molecules are collectively referred to as 3, but each water molecule is treated here as a separate fragment.
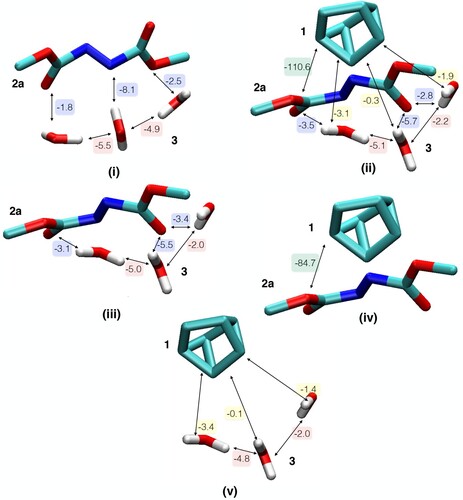
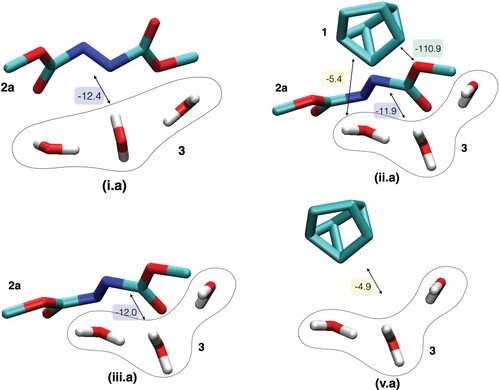
Figure 3. An alternative partitioning of the studied systems in Figure , but combining the water molecules in one fragment: (i.a) reactant conformation of 2a and 3, (ii.a) TS, (iii.a) TS without 1 and (v) TS without 2a. The CT between a given pair of fragments is indicated by an arrow and the corresponding value is given in kJ/mol.
In addition to ALMO, we also did an analysis of configurations i, ii, iii, iv and v using natural bond orbitals (NBO) [Citation18,Citation19]. The NBO computations were done at M06-2x/cc-pVDZ, as implemented in the Gaussian software [Citation16,Citation20]. The relevant NBO results are provided in Figure S2 in the ESI.
3. Results
The five initial configurations are illustrated in Figure with detailed values for CT, while the total results for the EDA of the five systems are provided in Table . The CT energies between each pair of 1, 2a and 3 are extracted from Figure and provided in Table . In all configurations in Figure , each molecule represents a fragment. Hence, the values in Table also includes interaction energy components between water molecules. The EDA results for the alternative partitioning, taking the water molecules as one fragment, are similarly provided in Figure and Tables and .
Table 1. EDA results in kJ/mol for the five configurations illustrated in Figure .
Table 2. CT (in kJ/mol) between the fragments in each of the five configurations illustrated in Figure .
Table 3. EDA results in kJ/mol for the four configurations illustrated in Figure .
Table 4. CT (in kJ/mol) between the fragments in each of the four configurations illustrated in Figure .
To establish a reference to compare the magnitudes of energy contributions, we did EDA for the 3 water molecules in the TS configuration at the same level of theory, but isolated from 1 and 2a. The values for frozen, polarisation and CT contributions are −7.6, −7.8 and −6.9 kJ/mol, respectively. As previously reported, frozen, polarisation and CT contributions are of equivalent importance in H-bonds [Citation12].
4. Discussion
As mentioned above, Jung and Marcus concluded that H-bonding is enhanced in the TS based on the expected electrostatics between the computed partial charges in both configurations. Interestingly, the frozen density interaction between 2a and 3 is more enhanced in i than in iii by −15 kJ/mol, see Table . Therefore, based on the electrostatic term alone, 2a is expected to form stronger H-bonds to interfacial waters in the reactant conformation than in the TS. This is an example of how conclusions based on population analysis could be misleading. However, water decreased the frozen density repulsion from 103 kJ/mol (between 1 and 2a in iv) to 78 kJ/mol (for the total system, ii). This decrease can be attributed to favourable frozen density attraction within water molecules (−7.6 kJ/mol for the isolated water trimer, see above) and through H-bonds to 2a (−18.5 kJ/mol for iii.a, see ).
We further analyse the remaining two terms that contribute to the H-bond strength. As summarised in Table , the CT between 2a and 3 is equivalent in both reactant (i) and TS (ii) configurations. The value is also equivalent when 2a and 3 are isolated from 1 in the TS configuration (iii). Clearly, the CT term does not indicate stronger H-bonding between 2a and 3 in the TS. The same conclusion is reached when the three water molecules are treated as one fragment, see Table . We actually notice additional CT between the water molecules and 1 which might contribute, albeit by a tiny fraction, to the energy lowering of the TS. More importantly, the presence of water molecules increases CT between the reactants ( 1 and 2a) in the TS configuration by around 30% (from −84.7 to −110.6 kJ/mol). To put it in perspective, this increase of −25.9 kJ/mol is more than three-fold larger than the aforementioned CT value in the isolated water trimer (−6.9 kJ/mol). The CT energy increase is associated with an increase of 61% of the total electron density transfer (from 184 to 297 millielectron, see Table S2 in the ESI).
The findings regarding the CT term are partially confirmed by the corresponding NBO calculations, see Figure S2 in the ESI. In agreement with ALMO EDA, the NBO analysis shows that the total CT energy between 2a and 3 is equivalent in all three configurations (i), (ii) and (iii). Nevertheless, according to the NBO results reported in Figure S2, there is only a marginal enhancement of CT between 1 and 2a, when the coordinates of water are included (an enhancement of 2.3 kcal/mol from −83.7 to −86.0 kcal/mol in the iv and ii configurations, respectively). This ambiguity can be attributed to the difference between CT in the NBO and ALMO EDA schemes. In NBO, the CT term is calculated from the off-diagonal blocks in the Fock-matrix of a given bond complex. This means that there are no variationally optimised intermediate states in the NBO scheme. Consequently, CT energy is not computed from a well-defined lowest-energy ‘zero-CT’ state as an accurate reference [Citation9,Citation21]. The ALMO constraint, however, strictly prohibits any CT between fragments at the optimised wavefunction, which is then considered to be the lowest-energy ‘zero-CT’ state [Citation13]. Therefore, the quantity
in Equation Equation3
(3)
(3) , accurately represents the energy of CT with no contribution from the polarisation energy [Citation9,Citation13,Citation21].
Besides amplifying the CT within reactants, the 3 water molecules increase the total polarisation of the system from −29.6 to −70.8 kJ/mol. This increase should not be falsely attributed to stronger H-bonding in the TS. The polarisation term for the isolated interaction between 2a and 3 in iii is equivalent to that in i, see Table . In addition, there are favourable polarisation interactions within water molecules (−7.8 kJ/mol, see above) and between 1 and 3 (−5.0 kJ/mol, see Table ). Hence, there seems to be no significant enhancement of the polarisation contribution to the extent of H-bonding in the TS as compared to the reactants.
5. Conclusions
Our results confirm that H-bonding to interfacial water molecules stabilises the TS of the cycloaddition of 1 and 2a. Nevertheless, factoring in all contributing terms to the strength of H-bonding, our EDA analysis shows that the H-bonds between the water molecules and 2a in both the reactant and TS configurations are of equivalent strengths, or even a bit stronger on the reactant side, contrary to what has been deduced by Jung and Marcus [Citation4] The energy lowering originates from the enhanced CT between 1 and 2a and the more favourable polarisation and frozen density terms for the whole system of 1, 2a and 3. This work illustrates the use of EDA to investigate the role of interfacial water in on-water catalysis. Further work is needed to reach more decisive conclusions. The configurations used in EDA should be properly sampled from a simulation of heterogeneous water–organic system. Reactants that were predicted to yield inefficient on-water catalysis, like 2c in Figure by Jung and Marcus [Citation4], should be compared to 2b and 2a. Last but not least, one should also consider a reaction mechanism involving proton-transfer as a possible reason for the observed kinetic isotope effect in on-water catalysis.
Supplemental Material
Download MS Word (7.5 KB)Acknowledgements
This research has been enabled via funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 716142). In addition, we acknowledge the allocation of computing resources by the Paderborn Center for Parallel Computing (PC2).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- S. Narayan, J. Muldoon, M. Finn, V.V. Fokin, H.C. Kolb and K.B. Sharpless, Angewandte Chemie Int. Edition 44 (21), 3275–3279 (2005). 10.1002/(ISSN)1521-3773 doi: 10.1002/anie.200462883
- R.N. Butler and A.G. Coyne, Org. Biomol. Chem. 14 (42), 9945–9960 (2016). doi:10.1039/C6OB01724J
- J.K. Beattie, C.S. McErlean and C.B. Phippen, Chem. A. Eur. J. 16 (30), 8972–8974 (2010). 10.1002/chem.v16:30 doi: 10.1002/chem.201001705
- Y. Jung and R. Marcus, J. Am. Chem. Soc. 129 (17), 5492–5502 (2007). doi:10.1021/ja068120f
- S. Mellouli, L. Bousekkine, A.B. Theberge and W.T. Huck, Angewandte Chemie Int. Edition 51 (32), 7981–7984 (2012). doi:10.1002/anie.v51.32 doi: 10.1002/anie.201200575
- L.R. Domingo, J.A. Saéz, R.J. Zaragozá and M. Arnó, J. Org. Chem. 73 (22), 8791–8799 (2008). doi:10.1021/jo801575g
- K. Karhan, R.Z. Khaliullin and T.D. Kühne, J. Chem. Phys. 141 (22), 22D528 (2014). doi:10.1063/1.4902537
- T.D. Kühne, M. Iannuzzi, M. Del Ben, V.V. Rybkin, P. Seewald, F. Stein, T. Laino, R.Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M.H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A.S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G.K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöß, M. Lass, I. Bethune, C.J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys. 152 (19), 194103 (2020). doi:10.1063/5.0007045
- M.J.S. Phipps, T. Fox, C.S. Tautermann and C.K. Skylaris, Chem. Soc. Rev. 44, 3177–3211 (2015). doi:10.1039/C4CS00375F
- L. Pecher and R. Tonner, WIREs Comput. Molecular Sci. 9 (4), e1401 (2019). doi:10.1002/wcms.1401
- R.Z. Khaliullin, E.A. Cobar, R.C. Lochan, A.T. Bell and M. Head-Gordon, J. Phys. Chem. A 111 (36), 8753–8765 (2007). doi:10.1021/jp073685z
- R.Z. Khaliullin, A.T. Bell and M. Head-Gordon, Chem. A Eur. J. 15 (4), 851–855 (2009). doi:10.1002/chem.200802107
- R.Z. Khaliullin and T.D. Kühne, Phys. Chem. Chem. Phys. 15, 15746–15766 (2013). doi:10.1039/c3cp51039e
- H. Elgabarty, R.Z. Khaliullin and T.D. Kühne, Nat. Commun. 6, 8318 (2015). doi:10.1038/ncomms9318
- Y. Zhao and D.G. Truhlar, Theor. Chem. Acc. 120 (1), 215–241 (2008). doi:10.1007/s00214-007-0310-x
- T.H. Dunning, J. Chem. Phys. 90 (2), 1007–1023 (1989). doi:10.1063/1.456153
- M. Krack and M. Parrinello, Phys. Chem. Chem. Phys. 2, 2105–2112 (2000). doi:10.1039/b001167n
- A.J. Foster and F. Weinhold, J. Am. Chem. Soc. 102 (24), 7211–7218 (1980). doi:10.1021/ja00544a007
- A.E. Reed, R.B. Weinstock and F. Weinhold, J. Chem. Phys. 83 (2), 735–746 (1985). doi:10.1063/1.449486
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, G.A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A.V. Marenich, J. Bloino, B.G. Janesko, R. Gomperts, B. Mennucci, H.P. Hratchian, J.V. Ortiz, A.F. Izmaylov, J.L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V.G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J.A. Montgomery, Jr. J. E. Peralta, F. Ogliaro, M.J. Bearpark, J.J. Heyd, E.N. Brothers, K.N. Kudin, V.N. Staroverov, T.A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A.P. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, J.M. Millam, M. Klene, C. Adamo, R. Cammi, J.W. Ochterski, R.L. Martin, K. Morokuma, O. Farkas, J.B. Foresman and D.J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016.
- Y. Mao, Q. Ge, P.R. Horn and M. Head-Gordon, J. Chem. Theory Comput. 14 (5), 2401–2417 (2018). doi:10.1021/acs.jctc.7b01256