
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Differential cross sections for the charge transfer reaction between Ar+ and CO have been measured using three-dimensional velocity map imaging in a crossed beam setup at the two relative collision energies 0.55 and 0.74 eV. We find dominant forward scattering with CO+ product ions predominantly in the vibrational levels =6,7 of the electronic ground state X
. This is indicative of a direct resonant mechanism for the two argon spin-orbit states. At both collision energies also an isotropic distribution with product ions exhibiting high internal excitation is observed. This is more pronounced at the higher collision energy, where the first electronically excited state A
becomes accessible. We conclude that the A-state is partially populated by the product ions at 0.74 eV collision energy and suggest that the isotropic distribution stems from the formation of a charge-transfer complex, in concurrence with previously performed studies.
GRAPHICAL ABSTRACT
1. Introduction
Ion neutral reactions play an important role in many environments, such as the ionosphere, the interstellar medium and in liquid phase chemistry. Charge transfer reactions are an important class of these reactions. They represent a simple reaction that occurs through the exchange of an electron, but can exhibit quite intricate mechanisms [Citation1,Citation2]. This becomes evident in a multitude of previously studied charge transfer reactions [Citation3–13]. One particularly interesting one is the reaction of Ar+ with the diatomic CO molecule; a reaction that has been studied for almost six decades [Citation13–37]. Especially early studies focused on kinetics, measuring rate constants and cross sections across a wide range of collision energies [Citation14–16,Citation18–20]. In these, the reaction rate was found to be well below the Langevin rate constant [Citation18,Citation35].
The potential energy curves for the product CO+ ion in the ground X and first electronically excited state A
are depicted in Figure with the vibrational energy levels given relative to the ground state CO+(X
,
=0). Ar+ exists in two possible spin-orbit states, namely the ground state Ar+
and the spin-orbit excited state Ar+
. The energy difference between the two states is 0.18 eV, as seen in Figure , and the title reaction is exoergic for both Ar+
and Ar+
by 1.75 eV and 1.92 eV, respectively. In experimental [Citation23,Citation29,Citation30] and theoretical [Citation28,Citation35] studies on absolute and relative cross sections, state-specificity in regard to the two different spin-orbit states of Ar+ has been shown. Although exact values differ there is a consensus that the
P
state exhibits higher reactivity than the
P
ground state. Despite the expected statistical population of 1:2 for Ar+ reactants, the excited state therefore contributes significantly to the resulting total cross sections. Furthermore, theory predicts the formation of CO+(A
) as the dominant product in the reaction with Ar+
, while Ar+
forms CO+ mostly in the electronic ground state X
[Citation28,Citation35].
Figure 1. Schematic representation of the potential energy curves of the CO+ ion in its electronic ground state X (lower curve) and first excited state A
(upper curve) with CO+(X
,
=0) as the point of reference. The data points are taken from Wei et al. [Citation39] and interpolation is done utilising cubic splines. The vibrational levels are calculated using the spectroscopic parameters from reference [Citation39]. Additionally, the energy levels of Ar+ in its two spin orbit states
and
are depicted together with vertical arrows illustrating the two collision energies 0.55 and 0.74 eV. For the two resonant levels CO+(X
,
=6) and CO+(X
,
=7) and the electronically excited level CO+(A
,
=0) the energies are given explicitly.
![Figure 1. Schematic representation of the potential energy curves of the CO+ ion in its electronic ground state X2Σ+ (lower curve) and first excited state A2Π+ (upper curve) with CO+(X2Σ+, ν′=0) as the point of reference. The data points are taken from Wei et al. [Citation39] and interpolation is done utilising cubic splines. The vibrational levels are calculated using the spectroscopic parameters from reference [Citation39]. Additionally, the energy levels of Ar+ in its two spin orbit states 2P3/2 and 2P1/2 are depicted together with vertical arrows illustrating the two collision energies 0.55 and 0.74 eV. For the two resonant levels CO+(X2Σ+, ν′=6) and CO+(X2Σ+, ν′=7) and the electronically excited level CO+(A2Π+, ν′=0) the energies are given explicitly.](/cms/asset/00a74082-cb2e-4d78-ae62-e9ceb9cfb0f2/tmph_a_1815885_f0001_oc.jpg)
Marx et al. [Citation24] performed the first product state resolved studies at thermal collision energies using an Ion Cyclotron Resonance (ICR) spectrometer. They found CO+(X,
=4) to be the predominant product. Two studies utilising laser-induced fluorescence at thermal and 0.2 eV collision energies by Hamilton et al. [Citation26] and Lin et al. [Citation25] found the vibrational energy distribution of the product CO+ to be centred around
=5. The discrepancy between these results was attributed to the underestimation of the thermal spread in the reagent velocities, especially in the ICR technique [Citation31]. However, the results could not be explained by either Franck-Condon factors, which are almost zero for vibrational levels higher than
[Citation28,Citation38], nor by charge resonance, which would favour the energetically closest lying
=6i,7ii Footnote1 levels (see Figure ). Parlant and Gislason [Citation35] calculated state-to-state cross sections at 1 eV collision energy finding the product CO+ ions predominantly in the electronic ground state and the vibrational levels
=1 and
=2 for the reactions with Ar+
and Ar+
, respectively, and no excitation above
=4. To account for the discrepancy with the experimental data, they proposed the formation of a long-lived charge-transfer complex at lower collision energies to explain the higher internal excitation of CO+. A recently published paper by He et al. [Citation13] measured differential cross sections of the reaction at 4.40, 6.40 and 8.39 eV collision energy. At all three energies they observed predominantly forward scattering and a clear preference for CO+ in the first electronically excited state.
We have obtained differential cross sections for the title reaction by using our ion-molecule crossed beam velocity map imaging setup at the two collision energies 0.55 and 0.74 eV, bridging the gap between earlier studies at thermal and the recent study at higher collision energies. This experimental method has proven to be capable of disentangling the internal product energy distribution in numerous charge transfer reactions [Citation3–13].
2. Methods
The experimental setup used for the crossed-beam experiment has been discussed previously in more detail [Citation40,Citation41]. To map the velocity distribution of the product ions, we employ a 3D variation of the velocity map imaging spectrometer, pioneered by Eppink and Parker [Citation42]. Ar+ ions are created by igniting a plasma in a molecular beam of neat argon, which is expected to produce a statistical distribution of 2:1 of the two different spin-orbit states Ar+ and Ar+
. Subsequently, the ions are accelerated and guided into an octupole radio-frequency ion trap, where they are thermalised to room temperature by addition of a buffer gas (in this case argon) to minimise the kinetic energy spread. After 40 ms trapping time, the ions are extracted and brought to the desired velocity by applying a potential difference between the trap and the VMI electrode stack. This allows us to vary the collision energy, while keeping the neutral beam velocity constant. The ion beam is then crossed at a 60
angle with the neutral beam of neat CO, produced by a supersonic expansion, at the centre of the VMI stack. The VMI extraction field is switched on at the appropriate time to accelerate the product ions perpendicular to the scattering plane. The ions hit a multi-channel plate (MCP) creating an electron avalanche, which is accelerated onto a phosphor screen. The position of impact is recorded by a CCD camera and the exact flight time by a photo-multiplier tube enabling the calculation of the three-dimensional velocity vector for each event. The kinetic energy, its spread, the angular and spatial distributions of both the ion and neutral beam (ionised by electron impact) can be measured utilising 2D velocity map imaging. By means of energy and momentum conservation, the internal energy distribution of the product ions is calculated. For this, single collision conditions have to be ensured. The velocity distributions are usually presented as a projection on a 2D-plane with respect to the velocity components parallel (
) and perpendicular (
) to the collision axis in the centre-of-mass frame.
3. Results
We have measured the differential cross sections for the charge-transfer reaction Ar+(P
) + CO in our crossed molecular beam setup using the VMI technique. In Figure (a,e) the velocity images of the product CO+ ion are shown at the two different collision energies (0.55 and 0.74 eV). The white and black solid rings represent the kinematic cutoff (maximum available kinetic energy) and the dashed inner rings the expected velocity of the CO+ product ions in the vibrational levels
= 6i,7ii. If the process resonantly couples to the closest reachable product state, the expected velocity distribution should peak around these dashed inner rings. Furthermore, the red ring in the velocity image for 0.74 eV collision energy corresponds to the energy needed to access the first electronically excited state A
, which is not accessible at the lower collision energy (see also Figure ).
Figure 2. Product ion velocity distribution (a, e), backward cut (b, f), internal energy (c, g) and angular distribution (d, h) at the two collision energies 0.55 (upper panel) and 0.74 eV (lower panel). Additionally, a Newton diagram depicting the collision in the centre-of-mass frame with θ as the scattering angle of the product CO+ is given at the top. The outermost solid rings in the velocity distributions give the kinematic cutoffs of CO+ for the reaction with Ar+ in its two spin-orbit states P
(white) and
P
(black). Furthermore, the dashed rings depict the expected velocity for CO+ ions in the vibrational levels
=6 and 7 for the reaction with the spin-orbit ground and excited state of Ar+, respectively. The red circles at the higher collision energy represent the energy level at which the first electronically excited state A
becomes accessible for the product CO+ ions.
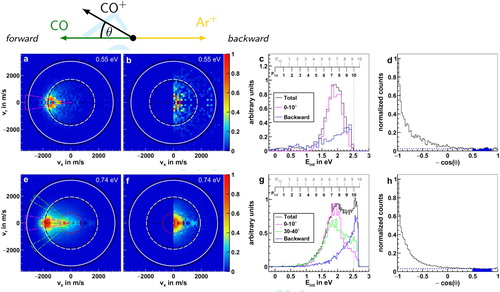
We find two features in the scattering images: dominant forward scattering, close to the expected velocity for resonant charge transfer, and an isotropically scattered contribution around zero velocity indicating high internal excitation. The main distribution in the scattering images coincides with the neutral beam vector in the centre-of-mass frame prior to collision, depicted by the arrows in forward direction in Figure (a,e), which clearly indicates that almost no momentum transfer takes place. From this we can conclude that all the energy released during the charge transfer reaction is converted into internal excitation. This is also evident in the internal energy distribution, which is given relative to the kinematic cutoff for Ar+(P
) + CO in Figure (c,g), since vibrational levels below
=3i,4ii are not significantly populated, which again implies the conversion of the released reaction energy into internal excitation of the molecular product instead of kinetic energy. The total internal energy distribution at 0.55 eV shows a single broad distribution centred slightly above the resonant energy levels
=6i,7ii. In contrast, we find an additional second contribution peaking at maximum internal excitation for 0.74 eV collision energy.
To gain further insight into the energy distributions of the forward scattered ions, we isolate them by considering a 20 cone in the scattering angle θ around the forward direction (magenta lines in Figure (a,e)). In the internal energy distribution of the lower collision energy (Figure (d)) this results in a comparable distribution to the complete one, while at the higher collision energy (Figure (g)) these selected ions contribute entirely to the first resonant peak at lower internal excitation. From Gaussian fits of the internal energy distribution of these cuts we obtain their FWHM of 0.61 and 0.65 eV at 0.55 and 0.74 eV collision energy, respectively. These are several times broader than the energy uncertainty stemming from the experiment, which we estimate to be approximately 100 meV [Citation40]. The maxima of the fits (1.93 and 1.87 eV) are positioned above the vibrational levels
6i,7ii of the resonant channel (1.81 eV, see Figure ). This implies the excitation of at least the next higher vibrational levels
7i,8ii, explaining the width and position of the internal energy distribution of the forward scattered ions. The excitation of rotational levels might also contribute to this shift.
At larger scattering angles in the forward direction we additionally observe the excitation of higher vibrational levels, visible in a cut from 30–40
in the internal energy distribution at 0.74 eV collision energy (green lines in Figure (g)). The majority of ions scattered into these higher angles, however, still populate the resonant states
6i,7ii. At the lower collision energy, we also observe excitation up to
=8i,9ii, but due to lower statistics, not much difference in the distribution can be seen when applying the same cut, which is therefore not shown.
The previously mentioned isotropic contribution to the images is best illustrated when only viewing the backward hemisphere of the scattering plane ( Figure (b,f)). Ions from this isotropic distribution exhibit high internal excitation, since they show velocities close to zero. Furthermore, in the internal energy distribution we observe a steady increase up to the highest available energy for the ions scattered into the backwards hemisphere (blue lines in Figure (c,g)). At 0.74 eV collision energy, these ions are responsible for the second peak at high internal excitation, while the contribution is not as strong at the lower collision energy, possibly due to the kinematic cutoff at 2.47 eV. From the energy diagram (Figure ) we know that only CO+ ions formed in the collision with Ar+(P
) have enough energy to contribute to this peak. Additionally, it can only originate from product ions in either the CO+(X
,
=10) or the CO+(A
,
=0) state, which are not individually resolved due to the energy uncertainty in our experiment.
Isotropic scattering around zero velocity is typical for a complex that dissociates after at least one rotational period [Citation37]. We classify it as an indirect mechanism. To quantify the ions formed in this indirect mechanism, we take a closer look at the angular distributions (Figure (d,h)). Apart from the clear peaks in forward direction, which stem from the dominant forward scattering, a baseline is evident across all angles. To determine the fraction of products under this baseline, we select an angular range from 120 to 150 (blue area), in order to exclude the contribution of the forward scattered ions, and extrapolate across the whole range (blue line). This gives us an upper bound for the relative amount of 23% for 0.55 and 33% for 0.74 eV collision energy, with a statistical error of under 1%.
4. Discussion
Earlier studies of the title reaction at thermal energy and at 0.2 eV found the product CO+ ion predominantly in the vibrational levels [Citation25,Citation26]. The results could not be explained by either Franck–Condon factors, which drop rapidly for
1 [Citation28,Citation38], or resonant charge transfer, which would favour the CO+(X
,
i,7ii) state. Therefore, the possibility of a long-lived charge-transfer complex was proposed [Citation26,Citation28,Citation33,Citation35,Citation37]. More recently differential cross sections were published by He et al. at higher collision energies [Citation13], observing primarily forward scattering into the A
state with high vibrational excitation. Some signal was also observed in the resonant channel CO+(X
,
6i,7ii), especially at the lowest collision energy of 4.44 eV.
In our experiments at 0.55 and 0.74 eV collision energy we find two mechanisms for this reaction. Direct, forward scattering into the vibrational levels around 6i,7ii and indirect, isotropic scattering with maximum internal excitation. Furthermore, there is very little momentum transfer in the direct mechanism, and we observe only small angular deflection indicative of large impact parameters. This process could also be identified by both measuring differential cross sections and calculating quasi-classical trajectories in the proton transfer reaction ArH++CO [Citation43]. Since the two states CO+(X
,
=6i,7ii) are the closest accessible ones to the reactant Ar+ in its two spin-orbital states, this suggests a resonant charge transfer as the dominant mechanism [Citation44]. This is similar to the reaction of Ar+ with H2 [Citation9], however no indirect part is observed there. As can be seen in the comparison of the 0–10
to the 30–40
cut ( Figure (c,g)), higher scattering angles lead to higher internal excitation, which was also observed for Ar++N2 [Citation7]. Higher scattering angles imply smaller impact parameters at which avoided crossings coupling to higher vibrational states might also contribute, as was suggested for the Ar++N2 reaction.
At 0.74 eV we see an increased relative amount of 33 % of isotropically scattered ions compared to 23% at 0.55 eV. These product ions exhibit a trend to higher internal excitation, as seen in Figure (c,g), suggesting the formation of a long-lived charge transfer complex (Ar-CO). Usually, an increase in collision energy decreases the stability of such a complex, since more energy needs to be distributed into internal degrees of freedom. Here however, we observe the opposite. Complex mediated charge transfer for this reaction has been proposed earlier, finding a preference for the product state distribution on the angle of attack [Citation26,Citation28,Citation33,Citation35,Citation37]. The enhancement of the indirect mechanism occurs at the energy where the A-state becomes accessible, supporting the hypothesis of an excited-state mediated charge transfer process. The energy resolution of our experiment is around 100 meV, therefore there is no clear way of differentiating between CO+(X
,
) and CO+(A
,
), which are separated by less than 1 meV. However, there is a clear preference towards the A-state at higher collision energies [Citation13], albeit in a direct process. With an increase in collision energy, a long-lived complex might no longer be stable, as suggested by Gislason et al. [Citation28], which would explain the lack of an isotropic distribution at higher collision energies as seen by He et al. Furthermore, the Franck–Condon factors for the lower vibrational levels of the A-state are significantly larger than those for the vibrationally excited levels of the electronic ground state [Citation28,Citation38].
Gislason et al. have shown, using a vibronic semiclassical method, that for CO+ ions formed in the reaction with Ar+(P
) there is a clear preference to populate the A-state while the opposite is the case for the reaction with Ar+(
P
) [Citation28]. Even though the ratio of Ar+(
P
) to Ar+(
P
) in the ion beam is 2:1, it has been shown both experimentally [Citation23,Citation29,Citation30] and theoretically [Citation28,Citation35] that the cross section of reactions with Ar+(
P
) is significantly higher. Kobayashi [Citation18] as well as Dotan and Lindinger [Citation22] measured reaction rates from thermal to a few eV collision energy. Initially, the rate decreases when increasing the collision energy, but at 0.8 eV it starts to increase again. This is exactly the collision energy needed to form CO+ in the first electronically excited state. Therefore, we expect that at least part of the ions with high internal excitation formed in the indirect mechanism at 0.74 eV populate the electronically excited A-state.
5. Conclusion
We have measured differential cross sections for the charge transfer reaction Ar+(P
) + CO(X
, ν=0)
Ar(
S
) + CO+(X
/A
,
) at the two collision energies 0.55 and 0.74 eV. At these energies the reaction is governed by forward scattering into vibrational levels of the ground state CO+ peaked around
=6i,7ii, which is attributed to a resonant charge transfer mechanism. CO+ products scattered at larger angles in forward direction evidence higher internal excitation. Further, an additional isotropic, low energy distribution is observed at 0.74 eV reactive collisions, indicative of a mechanism involving high product internal excitation. We attribute this partially to the formation of CO+ in the A
state. Further work with higher velocity resolution and state-of-the-art theory is needed to better resolve the ro-vibrational product state distribution and the role of the intermediate complex, also at higher collision energies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes
1 The superscripts i and ii are used to differentiate the vibrational levels accessed in the reaction with the different spin-orbit states Ar+ and Ar+
, respectively.
References
- T. Van Voorhis, T. Kowalczyk, B. Kaduk, L.P. Wang, C.L. Cheng, and Q. Wu, Annu. Rev. Phys. Chem. 61 (1), 149–170 (2010). doi:10.1146/annurev.physchem.012809.103324.
- S. Falcinelli, P. Candori, F. Pirani, and F. Vecchiocattivi, Phys. Chem. Chem. Phys. 19, 6933–6944 (2017). doi:10.1039/C7CP00614D.
- J. Mikosch, U. Frühling, S. Trippel, D. Schwalm, M. Weidemüller, and R. Wester, Phys. Chem. Chem. Phys. 8, 2990–2999 (2006). doi:10.1039/B603109A.
- L. Pei and J.M. Farrar, J. Chem. Phys. 136 (20), 204305 (2012). doi:10.1063/1.4719808.
- L. Pei and J.M. Farrar, J. Chem. Phys. 137 (15), 154312 (2012). doi:10.1063/1.4759265.
- L. Pei and J.M. Farrar, J. Chem. Phys. 138 (12), 124304 (2013). doi:10.1063/1.4796205.
- S. Trippel, M. Stei, J.A. Cox, and R. Wester, Phys. Rev. Lett. 110, 163201 (2013). doi:10.1103/PhysRevLett.110.163201.
- L. Pei, E. Carrascosa, N. Yang, S. Falcinelli, and J.M. Farrar, J. Phys. Chem. Lett. 6 (9), 1684–1689 (2015). doi:10.1021/acs.jpclett.5b00517.
- T. Michaelsen, B. Bastian, E. Carrascosa, J. Meyer, D.H. Parker, and R. Wester, J. Chem. Phys. 147 (1), 013940 (2017). doi:10.1063/1.4983305.
- J. Hu, C.X. Wu, Y. Ma, and S.X. Tian, J. Phys. Chem. A 122 (47), 9171–9176 (2018). doi:10.1021/acs.jpca.8b08005.
- C.X. Wu, J. Hu, M.M. He, and S.X. Tian, J. Phys. Chem. A 123 (40), 8536–8541 (2019). doi:10.1021/acs.jpca.9b06607.
- C.X. Wu, J. Hu, M.M. He, Y. Zhi, and S.X. Tian, Phys. Chem. Chem. Phys. 22, 4640–4646 (2020). doi:10.1039/C9CP06289K.
- M.M. He, J. Hu, C.X. Wu, Y. Zhi, and S.X. Tian, J. Phys. Chem. A 124 (17), 3358–3363 (2020). doi:10.1021/acs.jpca.0c02047.
- F.C. Fehsenfeld, E.E. Ferguson, and A.L. Schmeltekopf, J. Chem. Phys. 45 (1), 404–405 (1966). doi:10.1063/1.1727351.
- P. Warneck, J. Chem. Phys. 46 (2), 513–519 (1967). doi:10.1063/1.1840696.
- P.P. Ong and J.B. Hasted, J. Phys. B 2 (1), 91–101 (1969). doi:10.1088/0022-3700/2/1/313.
- M. Lipeles, J. Chem. Phys. 51 (3), 1252–1253 (1969). doi:10.1063/1.1672132.
- N. Kobayashi, J. Phys. Soc. Jpn. 36 (1), 259–266 (1974). doi:10.1143/JPSJ.36.259.
- A.F. Hedrick, T.F. Moran, K.J. McCann, and M.R. Flannery, J. Chem. Phys. 66 (1), 24–31 (1977). doi:10.1063/1.433673.
- C.J. Latimer, J. Phys. B 10 (3), 515–522 (1977). doi:10.1088/0022-3700/10/3/020.
- J. Danon and R. Marx, Chem. Phys. 68 (3), 255–260 (1982). doi:10.1016/0301-0104(82)87032-8.
- I. Dotan and W. Lindinger, J. Chem. Phys. 76 (10), 4972–4977 (1982). doi:10.1063/1.442843.
- T. Kato, K. Tanaka, and I. Koyano, J. Chem. Phys. 77 (1), 337–341 (1982). doi:10.1063/1.443610.
- R. Marx, G. Mauclaire, and R. Derai, Int. J. Mass Spectrom. Ion Phys. 47, 155–158 (1983). doi:10.1016/0020-7381(83)87159-9.
- G.H. Lin, J. Maier, and S.R. Leone, J. Chem. Phys. 82 (12), 5527–5535 (1985). doi:10.1063/1.448588.
- C.E. Hamilton, V.M. Bierbaum, and S.R. Leone, J. Chem. Phys. 83 (5), 2284–2292 (1985). doi:10.1063/1.449320.
- D.C. Parent, R. Derai, G. Mauclaire, M. Heninger, R. Marx, M.E. Rincon, A. O'Keefe, and M.T. Bowers, Chem. Phys. Lett. 117 (2), 127–131 (1985). doi:10.1016/0009-2614(85)85220-9.
- E.A. Gislason, G. Parlant, P. Archirel, and M. Sizun, Faraday Discuss. Chem. Soc. 84, 325–332 (1987). doi:10.1039/DC9878400325.
- B.G. Lindsay and C.J. Latimer, J. Phys. B: At. Mol. Opt. Phys. 21 (9), 1617–1625 (1988). doi:10.1088/0953-4075/21/9/019.
- G.D. Flesch and C.Y. Ng, J. Chem. Phys. 89 (5), 3381–3382 (1988). doi:10.1063/1.455708.
- D. Gerlich, J. Chem. Phys. 90 (1), 127–139 (1989). doi:10.1063/1.456518.
- C. Rebrion, B.R. Rowe, and J.B. Marquette, J. Chem. Phys. 91 (10), 6142–6147 (1989). doi:10.1063/1.457433.
- G. Parlant, P. Archirel, and E.A. Gislason, J. Chem. Phys. 92 (2), 1211–1220 (1990). doi:10.1063/1.458129.
- D.N. Vassallo and E.A. Gislason, Int. J. Mass Spectrom. Ion Proc. 110 (1), 83–92 (1991). doi:10.1016/0168-1176(91)80018-I.
- G. Parlant and E.A. Gislason, J. Phys. Chem. 97 (10), 2139–2146 (1993). doi:10.1021/j100112a013.
- I. Dotan and A.A. Viggiano, Chem. Phys. Lett. 209 (1), 67–71 (1993). doi:10.1016/0009-2614(93)87203-F.
- A.J. Midey and A.A. Viggiano, J. Chem. Phys. 109 (13), 5257–5263 (1998). doi:10.1063/1.477142.
- M.E. Wacks, J. Chem. Phys. 41 (4), 930–936 (1964). doi:10.1063/1.1726035.
- W. Xing, D. Shi, J. Zhang, J. Sun, and Z. Zhu, J. Quant. Spectrosc. Radiat. Transf. 210, 62–73 (2018). doi:10.1016/j.jqsrt.2018.02.008.
- R. Wester, Phys. Chem. Chem. Phys. 16, 396–405 (2014). doi:10.1039/C3CP53405G.
- M. Stei, E. Carrascosa, M.A. Kainz, A.H. Kelkar, J. Meyer, I. Szabó, G. Czakó, and R. Wester, Nat. Chem. 8 (2), 151–156 (2016). doi:10.1038/nchem.2400.
- A.T.J.B. Eppink and D.H. Parker, Rev. Sci. Instrum. 68 (9), 3477–3484 (1997). doi:10.1063/1.1148310.
- B. Bastian, E. Carrascosa, A. Kaiser, J. Meyer, T. Michaelsen, G. Czakó, W.L. Hase, and R. Wester, Int. J. Mass Spectrom. 438, 175–185 (2019). doi:10.1016/j.ijms.2018.12.004.
- E. Carrascosa, J. Meyer, and R. Wester, Chem. Soc. Rev. 46, 7498–7516 (2017). doi:10.1039/C7CS00623C.